Translate this page into:
H syndrome-Four new patients from India
2 Department of Rheumatology, Private Rheumatology Clinic, Mumbai, Maharashtra, India
3 Department of Genetic Medicine, Deenanath Mangeshkar Hospital and Research Center, Pune, Maharashtra, India
4 Department of Pediatrics, Kasturba Medical College, Manipal University, Mangalore, Karnataka, India
Correspondence Address:
Abraham Zlotogorski
Department of Dermatology, Hadassah Hebrew University Medical Center, PO Box 12000, Jerusalem - 9112001
Israel
| How to cite this article: Molho-Pessach V, Varma M, Godbole K, Kamath N, Zlotogorski A. H syndrome-Four new patients from India . Indian J Dermatol Venereol Leprol 2014;80:579 |
Sir,
H syndrome (OMIM 602782) is an autosomal recessive disorder characterized by cutaneous hyperpigmentation, hypertrichosis, hepatosplenomegaly, hearing loss, heart anomalies, hypogonadism, low height (short stature), hyperglycemia/diabetes mellitus and hallux valgus/flexion contractures. [1],[2] It is caused by biallelic mutations in the SLC29A3 gene encoding the human equilibrative nucleoside transporter 3. [2] Recently, the clinical, histopathological and molecular findings in 79 patients with H syndrome have been summarized. [3] Numerous other clinical features have been recognized to be part of the wide phenotypic spectrum including massive lymphadenopathy, pancreatic exocrine insufficiency, various heart and renal abnormalities, bone sclerosis, lytic bone lesions, swollen respiratory mucosa, infiltrated cheeks, and recurrent fever. [3]
A pathognomonic finding, the most common clinical feature, seen in 68% of patients is cutaneous hyperpigmentation accompanied by induration and hypertrichosis, which involves the medial thighs and shins with sparing of the knees. Skin biopsies show a dense inflammatory dermal and subcutaneous infiltrate composed mainly of CD68-positive and at times also S100-positive histiocytes, which is eventually replaced by fibrosis. [4] This histological feature implies that H syndrome is a form of inherited histiocytosis.
We present four newly diagnosed Indian patients with H syndrome originating from three families.
Case 1
A 6-year-old Indian boy, born of a non-consanguineous marriage, developed weekly febrile episodes at the age of 6 months, lasting 2 to 3 days, accompanied by swelling and tenderness of dorsal feet. As he began walking, he complained of pain after a few minutes of walking. The swelling and pain had a waxing and waning course over the following years. Hyperpigmentation with overlying hypertrichosis involving the trunk and lower limbs appeared at the age of 18 months. The patient also suffered from hearing loss.
Physical examination revealed well-defined, hyperpigmented, indurated plaques with hypertrichosis over the lower limbs [Figure - 1]. Left orbital swelling and mildly swollen cheeks were also seen. Eye examination showed mild conjunctival injection and arcus senilis. Cervical and inguinal lymph nodes were bilaterally enlarged. He had a small penis (micropenis). Laboratory evaluation revealed microcytic anemia, elevated liver function tests, markedly increased erythrocyte sedimentation rate (ESR) and C-reactive protein and elevated triglycerides (374 mg/dL, normal range 26-123 mg/dL). Abdominal ultrasound showed mild hepatosplenomegaly. Magnetic resonance imaging of the thighs showed extensive bilateral inguinal and iliac lymphadenopathy, as well as diffuse hyperintensity of the subcutaneous tissue and the lower third of the thigh muscles of both limbs.
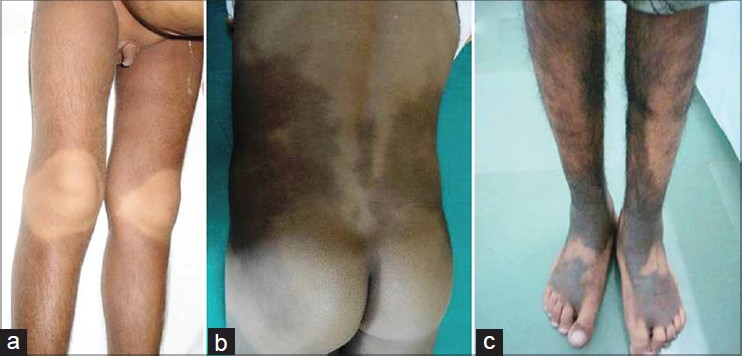 |
| Figure 1: Typical pattern of cutaneous hyperpigmentation and hypertrichosis: (a) Hyperpigmentation, hypertrichosis of lower limbs with sparing of knees in the patient from family 1. Micropenis is also seen. (b) Hyperpigmentation of the lower back, sparing of the buttocks, in the patient from family 2. (c) Hyperpigmentation and hypertrichosis of shins and dorsum of feet in the older patient from family 3 |
Case 2
A 16-year-old Indian female was referred due to short stature, sensorineural hearing loss, insulin dependent diabetes mellitus, delayed puberty and diffuse cutaneous hyperpigmentation and hypertrichosis. On physical examination, the patient was noted to have indurated, hyperpigmented patches with accompanying hypertrichosis overlying the trunk, dorsum of feet, shins and inner thighs with sparing of the knees and buttocks [Figure - 1]. Additional findings were hepatosplenomegaly, dilated lateral scleral vessels, arcus senilis, infiltrated cheeks, genital masses, hallux valgus and lateral tibial torsion. Laboratory evaluation revealed mild microcytic anemia, highly elevated ESR and hypogonadotropic hypogonadism.
Cases 3 and 4
Two Indian brothers, 19-year-old and 12-year-old, born to remotely related cousins presented with progressive cutaneous hyperpigmentation and hypertrichosis. The elder brother suffered from bilateral sensor-neural hearing loss. During childhood he was diagnosed with idiopathic thrombocytopenic purpura. On physical examination, he was noted to have widespread hyperpigmented patches with hypertrichosis over the lower limbs, abdomen, lower back and upper extremities [Figure - 1]. Dilated lateral scleral vessels, swollen cheeks and gynecomastia were also observed. His younger brother had similar cutaneous findings and also suffered from bilateral sensor-neural hearing loss.
The patients presented here share the unique cutaneous hyperpigmentation accompanied by sclerodermatous thickening and hypertrichosis of the lower limbs, in a bilateral, symmetric distribution with typical sparing of the knees. This pathognomonic clinical feature of H syndrome [3] should ideally be confirmed by mutation analysis of SLC29A3. However, in certain instances, DNA analysis is difficult to obtain and yet the diagnosis can be confidently made, especially when other common features of the disorder are present, such as hearing loss, short stature and/or camptodactyly.
We were able to find published reports of a total of nine Indian patients, including the four above, who have been diagnosed with H syndrome. [3],[5] With the inclusion of the patients in this report, as well as the 80 patients described with molecular confirmation [3],[5] and 12 patients without such analysis, [3] there are published reports of 96 patients with H syndrome. This is a remarkable number of patients for a relatively novel autosomal recessive disorder implying that the disorder is not rare, especially in populations where consanguinity is common. Increased awareness among dermatologists about the pathognomonic cutaneous phenotype may lead to the identification of other patients.
ACKNOWLEDGMENTS
We thank the family members for their participation in this study. This study was supported in part by The Authority for Research and Development, Hebrew University of Jerusalem (Dr. Zlotogorski) and the Hadassah-Hebrew University Joint Research Fund (Dr. Molho-Pessach).
| 1. |
Molho-Pessach V, Agha Z, Aamar S, Glaser B, Doviner V, Hiller N, et al. The H syndrome: A genodermatosis characterized by indurated, hyperpigmented, and hypertrichotic skin with systemic manifestations. J Am Acad Dermatol 2008;59:79-85.
[Google Scholar]
|
| 2. |
Molho-Pessach V, Lerer I, Abeliovich D, Agha Z, Abu Libdeh A, Broshtilova V, et al. The H syndrome is caused by mutations in the nucleoside transporter hENT3. Am J Hum Genet 2008;83:529-34.
[Google Scholar]
|
| 3. |
Molho-Pessach V, Ramot Y, Camille F, Doviner V, Babay S, Luis SJ, et al. H syndrome: The first 79 patients. J Am Acad Dermatol 2014;70:80-8.
[Google Scholar]
|
| 4. |
Colmenero I, Molho-Pessach V, Torrelo A, Zlotogorski A, Requena L. Emperipolesis: An additional common histopathologic finding in H syndrome and Rosai-Dorfman disease. Am J Dermatopathol 2012;34:315-20.
[Google Scholar]
|
| 5. |
Mohanan S, Chandrashekar L, Semple RK, Thappa DM, Rajesh NG, Negi VS, et al. H syndrome with a novel homozygous R134C mutation in SLC29A3 gene. Int J Dermatol 2013;52:820-3.
[Google Scholar]
|
Fulltext Views
1,952
PDF downloads
772





![[Figure - 1]](#fig_ijdvl_2014_80_6_579_144229_u1.jpg){kind=link}