Translate this page into:
Hereditary spherocytosis presenting as indolent leg ulcers
Correspondence Address:
K Muhammed
Department of Dermatology and Venereology, Medical College, Calicut
India
| How to cite this article: Muhammed K, Lilly S, Shereef P H. Hereditary spherocytosis presenting as indolent leg ulcers. Indian J Dermatol Venereol Leprol 1997;63:55-57 |
Abstract
Indolent leg ulcertation, which is the rarest manifestation of hereditary spherocytosis, started at the age of 5 years affecting a 15-year-old boy and his mother is reported. Review of literature showed very few reports from India and abroad. The response to oral folic acid was excellent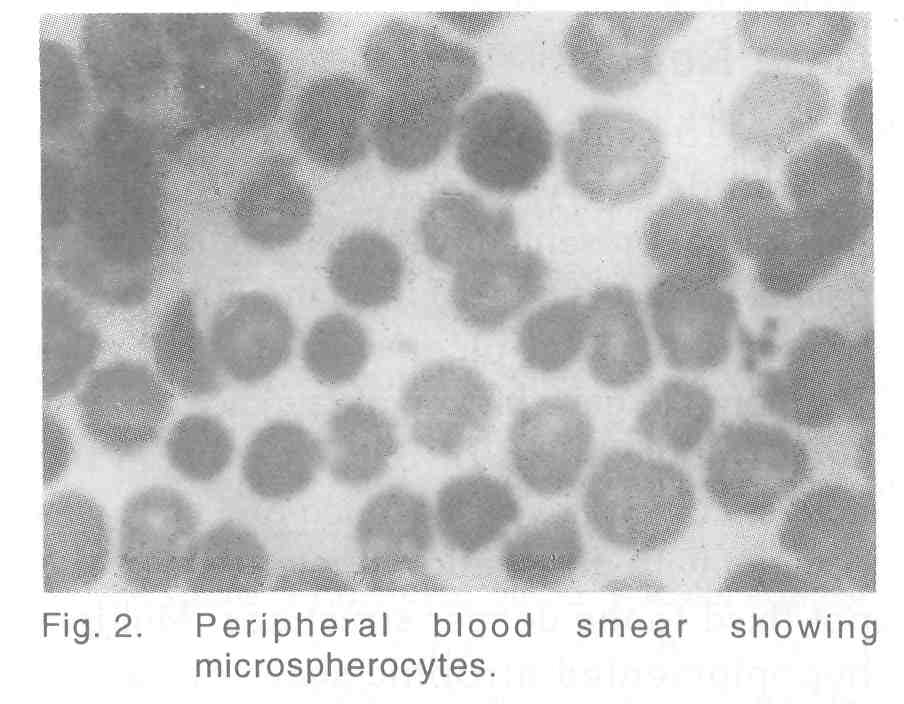 |
|
 |
|
Introduction
Hereditary spherocytosis (HS), a rare red cell hemolytic disorder, is usually transmitted as autosomal dominant inheritance in 75% of cases. But in the rest of cases it can be autosomal recessive or autosomal dominant with reduced penetrance or mutations, especially when there is no family history of the disease. The pathophysiology is the defect in the cytoskelecton of red blood cells (RBC). This defect is in the protein skeleton of RBC membrane in the form of marked deficiency of spectrin synthesis or synthesis of unstable spectrin.[1] Functionally abnormal spectrin with defective binding to protein 4.1 has been described, which is responsible for defective spectrin-actin interaction, which in turn is responsible for membrane instability and hemolytic disease. Loss of surface area is the fundamental characteristic of HS cells, leading to decreased ratio of surface area to volume. An intact spleen is an extrinsic prerequisite for hemolysis. Moderate anaemia, intermittent jaundice and splenomegaly, singly or together are the clinical features of HS. Anaemia or hyperbilirubinaemia may be of such magnitude as to require exchange transfusion in neonates. Usual complications are gall stones of pigmentary type, hemochromatosis or skeletal abnormalities.[1]An unusual complication is chronic leg ulceration. Chronic dermatosis and pigmentation may be the only evidence of healed ulcers.
We report HS affecting two generations of a family presenting as chronic leg ulcers, started at the age of 5 years in the patient and his mother. To the best of our knowledge, only one case of HS with leg ulcer has been reported from India.[2]
Case Report
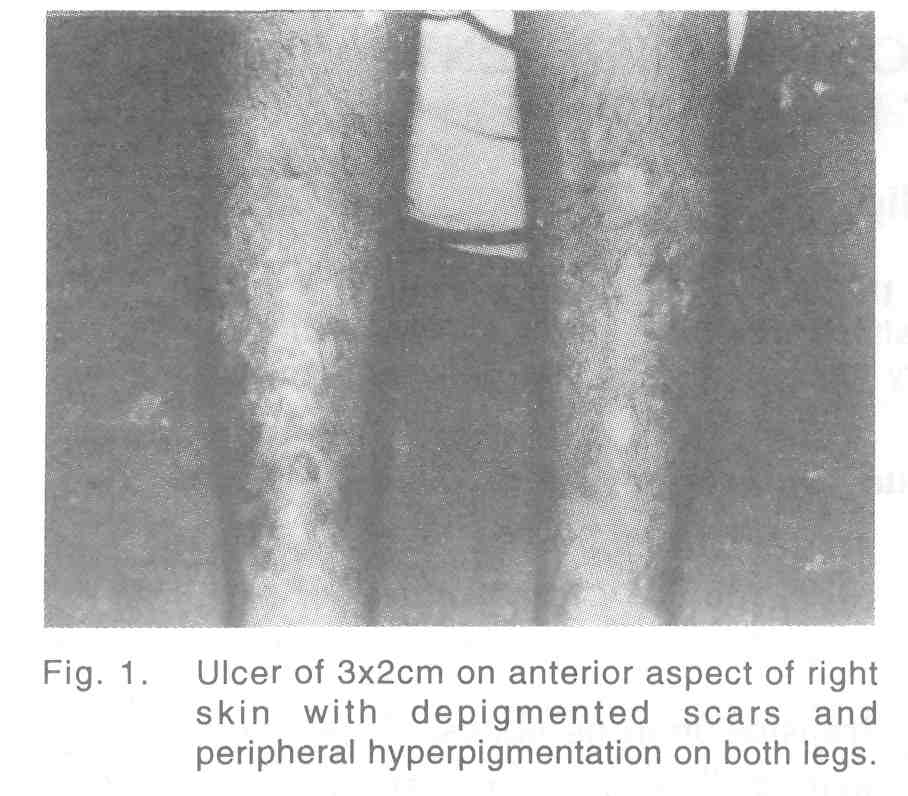
A 15-year-old boy, born of a full term normal devivery of non-consanguineous parentage, presented with bilateral recurrent painful leg ulcers [Figure - 1] from the fifth year of life. The present ulcer was of 3 months duration. He gave history of jaundice 8 years ago. The ulcer was of 3x2 cm size on the right shin, margins well defined and floor was covered with granulation tissue, tender and not fixed to the deeper structures. Multiple hypopigmented atrophic scars of healed ulcers with peripheral hyperpigmentation were seen on both legs. No significant inguinal lymphadenopathy or varicose veins were detected. Splenomegaly was present, 3cm below left costal margin. Baseline hematological examinations were within normal limits except mild anaemia (hemoglobin level 12gm/dl). Urinary urobilinogen was increased, but bile salts and bile pigments were absent. Liver function test showed a marked elevation of unconjugated bilirubin (7.6mg/dl). Peripheral smear examination revealed multiple densely staining small spherical RBCs (microspherocytes and polychromatophilia) [Figure - 2]. Direct Coomb′s test and sickling test were negative. Reticulocyte count and osmotic fragility test were markedly increased (lysis began in 0.56% and completed in 0.16% saline). Hemoglobin electrophoresis was negative for HbS or HbA2. Roentgenographic bone survey was normal. Ultrasonogram of abdomen revealed splenomegaly. Pus culture from the ulcer revealed Staph aureus sensitive to cloxacillin and cefazolin.
Screeing of family members was done. His 45-year-old mother were icteric and with enlarged spleen. She gave history of recurrent indolent leg ulcers, since the age of 5 years as evidenced by the presence of multiple atrophic scars on the shins.
The patient was treated with 5mg folic acid daily and cloxacillin 500mg 6 hourly with cleaning and dressing of the ulcer. Ulcers healed within a period of 1 month. The patient was continued with 5mg folic acid daily and he is being followed up regularly.
Discussion
The first case of leg ulcer in HS was reported in 1902 by Barlow and Shwa.[3] Leg ulceration is common in sickle cell disease and thalassemia, but very few cases are reported in HS. Taylor (1939) reported a case of chronic leg ulcer in HS.[4] In our case both the patient′s and his mother′s main problem was recurrent leg ulceration, that too started at the age of 5 years. This type of presentation is reported rarely. The anemia was refractory to treatment with iron preparation but responded well to oral folic acid and ulcers healed. The exact cause of leg ulceration is unknown. As in any case of chronic anemia, the lack of oxygenation and trauma may be a cause, along with the rheological disturbances.[5] In HS hemolytic, aplastic or megaloblastic crisis can occur associated with stress, pregnancy or infections especially viral. Splenectomy is the treatment of choice done after 6 years of age.
This case is being reported to bring to the notice of the dermatologists a rare but often overlooked cause for indolent leg ulcers.
| 1. |
Lukens JN. Hereditary spherocytosis and other hemolytic anemias associated with abnormalities of red blood cell membranes and cytoskeleton. In:Winthrobe's clinical hematology. Philadelphia:Lea and Febiger, 1993:965-72.
[Google Scholar]
|
| 2. |
Dash S, Dash RJ, Mahajan KK, et al. Bilateral chronic leg ulcers in Hereditary spherocytosis. Ind J Dermatol Venereol Leprol 1981;4:317-9.
[Google Scholar]
|
| 3. |
Barlaw T, Shaw HB. Inheritance of recurrent attacks of jaundice of abdominal crisis with hepatosplenomegaly. Tr Clin Soc London 1902;35:155-61 (quoted in 2).
[Google Scholar]
|
| 4. |
Taylor ES. Chronic ulcer of the leg associated with congenital hemolytic jaundice. JAMA 1939;112:1574.
[Google Scholar]
|
| 5. |
Vancheidt W, Leder D, Vanscheidt E, et al. Leg ulcers in a patient with spherocytosis-a clinicopathological report. Dermatologica 1990;181:56-9.
[Google Scholar]
|





![[Figure - 1]](#fig_ijdvl_1997_63_1_55_4508_1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_1997_63_1_55_4508_2.jpg){kind=link}