
Translate this page into:
Homozygous familial hypercholesterolemia with xanthomas and a recurrent mutation
Corresponding author: Dr. Vinod Scaria, Department of Genomics, CSIR-Institute of Genomics and Integrative Biology, Sukhdev Vihar, New Delhi, India. vinods@igib.in
-
Received: ,
Accepted: ,
How to cite this article: Raman T, Imran M, Agarwal M, Singh S, Bhoyar RC, Gupta A, et al. Homozygous familial hypercholesterolemia with xanthomas and a recurrent mutation. Indian J Dermatol Venereol Leprol. 2025;91:S71-S73. doi: 10.25259/IJDVL_980_2023
Dear Editor,
Familial hypercholesterolemia (FH) is an autosomal co-dominant disorder characterised by a lifelong elevation of LDL cholesterol (LDL-C). It is caused by pathogenic mutations in the low-density lipoprotein receptor (LDLR), apolipoprotein B (APOB), or proprotein convertase subtilisin/ kexin type 9 (PCSK9) genes. FH manifests in two major clinical forms: heterozygous FH (1 in 313 people; LDL-C > 190 mg/dL) and homozygous FH (1 in 400,000 people; LDL-C levels > 500 mg/dL).1 Clinically, the presence of widely distributed and large-sized xanthomas signifies severe and long-term FH, commonly observed in homozygous FH.
A 35-year-old male presented with multiple, gradually enlarging, firm, non-tender, skin-coloured, and hyperpigmented papulonodules symmetrically distributed over bony prominences for 20 years [Figures 1a–1c]. The lesions ranged in size from 0.5 × 0.5 × 0.5 cm over the hands to 6 × 5 × 3 cm over the buttocks. In addition, the patient reported intermittent chest and joint pain during periods of physical effort. There was a history of similar lesions over the elbows of his paternal grandfather. Dermoscopy of the lesion over the left cubital fossa revealed a yellow-brown structureless area. [Figure 2a]. The fasting lipid profile was deranged: total cholesterol (TC): 500 mg/dL (reference range <200 mg/dL); triglyceride 302 mg/dL (reference range < 150 mg/dL); LDL > 300 mg/dL (reference range <100 mg/dL); VLDL: 60.4 mg/dL (reference range: 5–40 mg/dL); high density lipoprotein (HDL): 21 mg/dL (reference range: 40–60 mg/dL). Other biochemical and haematological investigations were normal. Skin biopsy from a tuberous xanthoma demonstrated a collection of fat-laden histiocytes with bubbly cytoplasm in the dermis [Figure 2b].
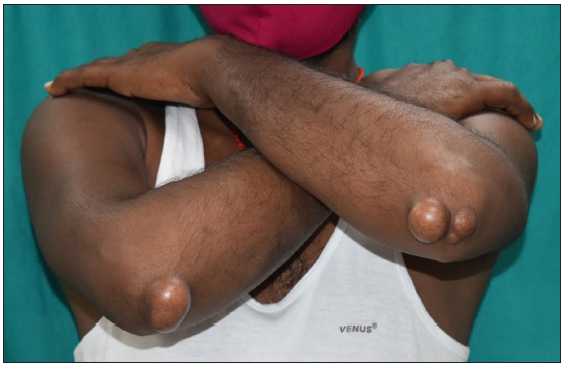
- Tuberous xanthomas present over both the elbows.
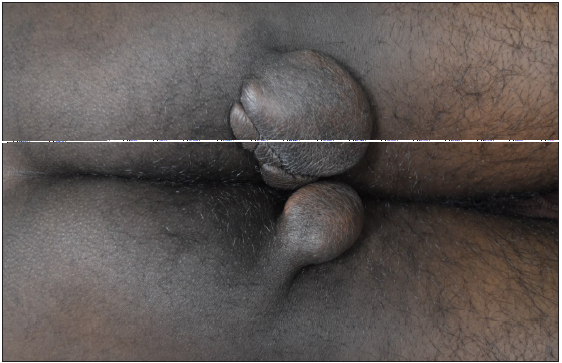
- Tuberous xanthomas over the buttocks.
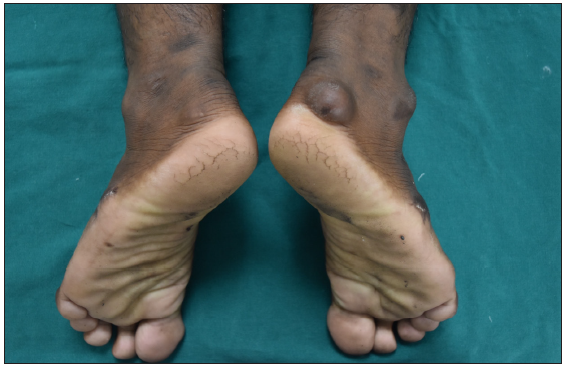
- Tendinous xanthoma over the right ankle.
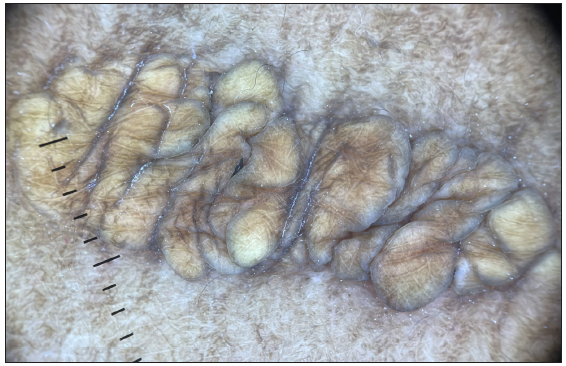
- Dermoscopy of the xanthoma over the left cubital fossa revealed a yellow-brown structureless area (Dermlite 4, 10x magnification, non-polarised).
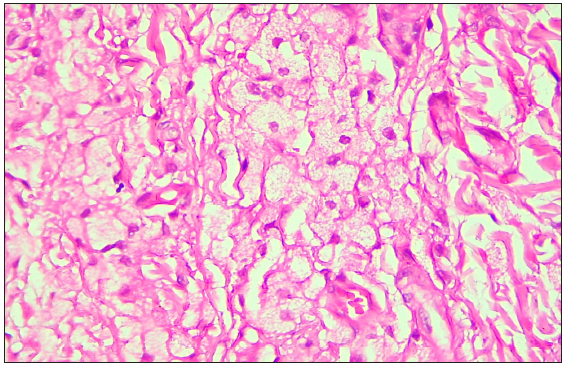
- Photomicrograph demonstrated collection of fat-laden histiocytes with bubbly cytoplasm in the dermis. (Haematoxylin and Eosin, 100x)
Next-generation sequencing was performed on the proband and family members, specifically targeting the exons of LDLR, APOB, and PCSK9 genes. Additionally, the analysis included a screening of pharmacogenomics variants rs2306283, rs4149056, and rs2231142, which are relevant to statin-associated musculoskeletal symptoms (SAMS).2 The proband was found to be homozygous for the c.530C>T (p.S177L) mutation in exon 4 of the LDLR gene, while his mother carried the mutation in a heterozygous state [Figures 3a–3c]. His younger brother tested negative for the mutation. Upon analysing the pharmacogenomic variants, we found that the proband and mother were heterozygous for the rs2306283 (c.388A>G) and rs4149056 (c.521T>C) variants in SLCO1B1 gene, leading to the SLCO1B1 decreased function phenotype necessitating no change in the treatment that had been prescribed to the patient (atorvastatin 40 mg and ezetimibe 10 mg) for the management of intermittent chest pain and hypercholesterolemia as they complied with the recent Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines.2
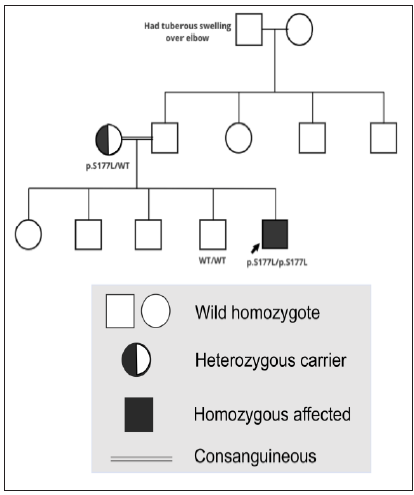
- Pedigree of the family.
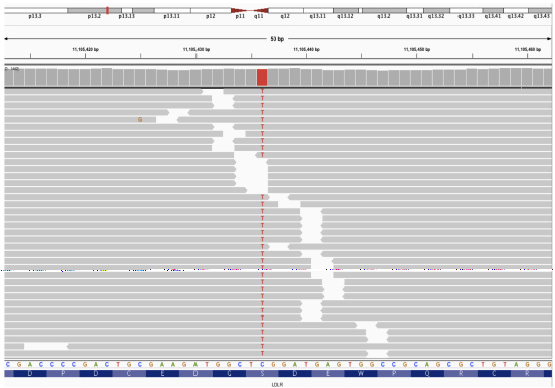
- Integrative Genomics Viewer (IGV) screenshot of the mutation in proband (homozygous).
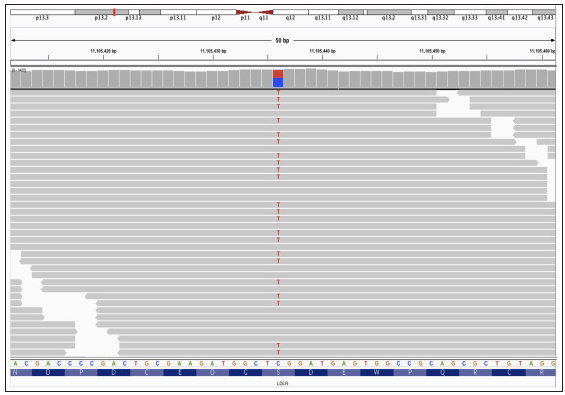
- Integrative Genomics Viewer (IGV) screenshot of the mutation in proband’s mother (heterozygous).
We present a consanguineous family from Pune, Maharashtra, with FH harbouring the LDLR: c.530C>T (p.S177L) variant. This disease-specific mutation, pathogenic mutation 1 of the moderate pathogenic criteria as per the American College of Medical Genetics and Genomics (ACMG)/American Association of Molecular Pathology (AMP) variant classification has been previously reported in three patients from India, including a heterozygous case from Mumbai, Maharashtra, and two homozygous cases from Kerala and Orissa.3–5 The heterozygous case had a possible diagnosis of FH according to the Dutch Lipid Clinic Network (DLCN) criteria, with marginally high LDL-C (112 mg/dL). In contrast, the two homozygous cases showed severe hypercholesterolemia, with LDL-C levels of 626 and 873 mg/dL, respectively. Both patients were from non-consanguineous families and presented with tendon xanthomata and corneal arcus.3,4 The observation of this mutation in Indian patients previously suggests its likely widespread occurrence within the Indian population. The South Asian population has been found to have the highest allele frequency for this mutation in gnomAD (v2.1.1), at 0.00006533 (2 individuals), followed by the East Asian (1 individual) and the non-Finnish European population (1 individual).6 Furthermore, the mutation has been reported in several patients from different countries, including Spain, Portugal, and Brazil.7
Additionally, the finding of pharmacogenomic variants in the affected individuals highlights the potential importance of considering pharmacogenomics in the management of patients receiving statin therapy. In this case, the patient had a SLCO1B1 decreased function phenotype and received combination therapy of atorvastatin 40 mg and ezetimibe 10 mg as per the CPIC guidelines without experiencing any adverse effects.2 It is essential to consider specific phenotypes, such as the SLCO1B1 poor function phenotype, in which the use of more than 20 mg of atorvastatin may not be advisable.2 Screening patients for these variants before initiating treatment enables healthcare providers to stratify individuals at a higher risk of developing myopathy, thereby facilitating the implementation of personalised approaches to optimise patient outcomes and promote treatment adherence.
The widespread occurrence of the LDLR: c.530C>T (p.S177L) variant in diverse populations and its functional impact on LDL receptor activity are consistent with its pathogenic role in FH. The identification of this mutation in our Indian family with FH adds to the growing evidence of its significance in the context of hypercholesterolemia worldwide. Clinicians should consider next-generation sequencing as a diagnostic tool for the early detection of FH. Additionally, screening of family members and at-risk individuals should be instructed to initiate early therapy and avert significant adverse events.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Worldwide prevalence of familial hypercholesterolemia: Meta-analyses of 11 million subjects. J Am Coll Cardiol. 2020;75:2553-66.
- [CrossRef] [PubMed] [Google Scholar]
- The clinical pharmacogenetics implementation consortium guideline for SLCO1B1, ABCG2, and CYP2C9 genotypes and statin-associated musculoskeletal symptoms. Clin Pharmacol Ther. 2022;111:1007-21.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Spectrum of mutations in homozygous familial hypercholesterolemia in India, with four novel mutations. Atherosclerosis. 2016;255:31-6.
- [CrossRef] [PubMed] [Google Scholar]
- Genetic analysis of familial hypercholesterolemia in Asian Indians: A single-center study. J Clin Lipidol. 2020;14:35-45.
- [CrossRef] [PubMed] [Google Scholar]
- Screening of PCSK9 and LDLR genetic variants in Familial Hypercholesterolemia (FH) patients in India. J Hum Genet. 2021;66:983-93.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434-43.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Clinical and molecular aspects of familial hypercholesterolemia in Ibero-American countries. J Clin Lipidol. 2017;11:160-6.
- [CrossRef] [PubMed] [Google Scholar]




