Translate this page into:
Identification of a novel missense mutation in the NOD2 gene in a Chinese child with early-onset sarcoidosis
2 Department of Dermatology, Institute of Plastic Surgery, Chinese Academy of Medical Sciences and Peking Union Medical College, Beijing, China
Correspondence Address:
Jianfang Sun
Department of Pathology, Institute of Dermatology, Chinese Academy of Medical Sciences and Peking Union Medical College, 12 Jiangwangmiao Road, Nanjing, Jiangsu 210042
China
| How to cite this article: Wang X, Zhang X, Zhang W, Sun J. Identification of a novel missense mutation in the NOD2 gene in a Chinese child with early-onset sarcoidosis. Indian J Dermatol Venereol Leprol 2018;84:645 |
Sir,
Early-onset sarcoidosis is known to appear in children younger than 4 years of age, and is characterized by a distinct triad of skin, joint and eye disorders without pulmonary involvement. Blau syndrome has been identified as the familial phenotype of this type of granulomatous autoinflammatory disease. Both are monogenic syndromes caused by mutations in the NOD2 gene.[1] We found a novel p. Y563D mutation in NOD2 gene in a Chinese boy with early-onset sarcoidosis.
A 6-year-old Chinese boy presented to the Institute of Dermatology, Chinese Academy of Medical Sciences and Peking Union Medical College with subcutaneous nodules over the joints and multiple asymptomatic papules over the trunk and limbs. He had developed subcutaneous nodules over the ankle and wrist joints, which was accompanied by symmetric polyarthritis affecting the wrist, ankle and proximal interphalangeal joints, two years back. He also developed multiple non-pruritic papules over the trunk and limbs, six months ago. His medical history was normal except for cleft lip and palate, which were repaired at 10 months of age. No familial history of similar disease was present. On dermatological examination, numerous 1–2 mm sized, red flat-topped papules covered the trunk and extremities [Figure - 1]a and b]. Coexisting subcutaneous nodules were found over the affected joints and the dorsum of hand and foot [Figure - 1]c and d]. Camptodactyly of the fingers was observed. No abnormalities were detected in ophthalmological examinations. Routine blood tests, serum calcium level, hepatic function panel and urine analysis tests showed normal results. The serum angiotensin-converting enzyme was 135 U/L (reference range, 20–100 U/L). A tuberculin test was negative. Chest radiographs were unremarkable. Radiographs of the involved joints revealed swollen soft tissue, with no bone abnormalities. Skin biopsy demonstrated noncaseating granulomas admixed with scant lymphocytes infiltration [Figure - 1]e. Polarized microscopy failed to reveal foreign material. Periodic acid-Schiff and Fite's staining were negative for organisms.
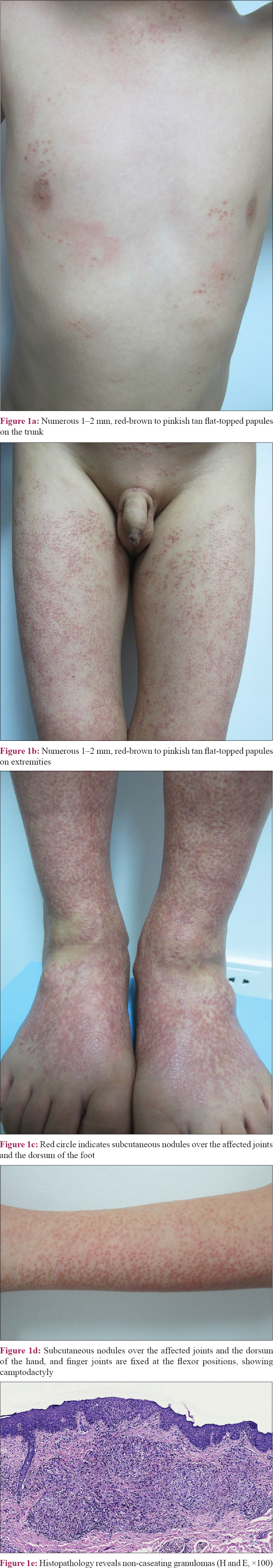 |
| Figure 1 |
Genetic analysis via direct sequencing of the NOD2 gene revealed a heterozygous missense mutation in c. 1687 T>G (p. Y563D) [Figure - 2], which was not detected in his unaffected parents, and a heterozygous synonymous mutation c. 1761 T>G (p. R587R), which was inherited from his father [Figure - 3]. The experimental program Polymorphism phenotyping v2 (http://genetics.bwh.harvard.edu/pph2/) predicted that this missense mutation is “probably damaging” with a score of 0.999.
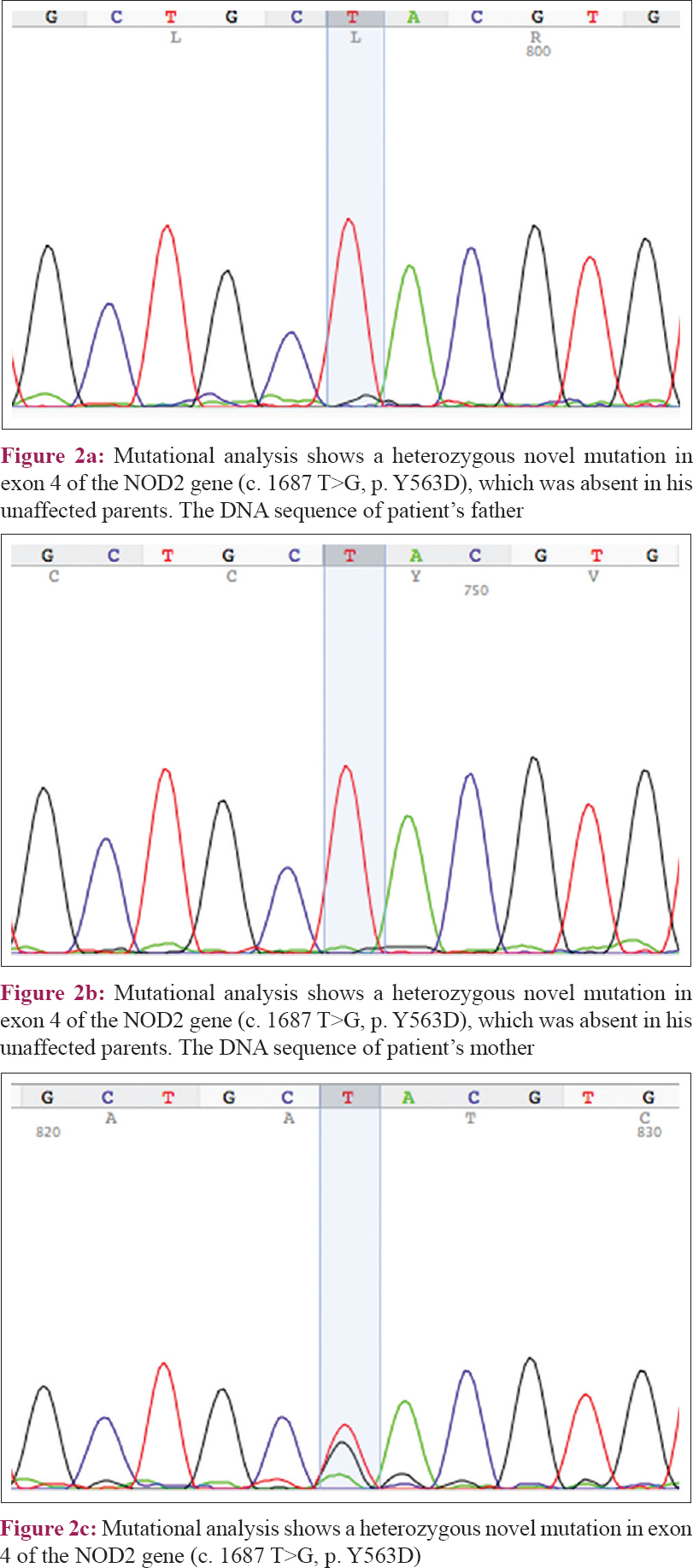 |
| Figure 2 |
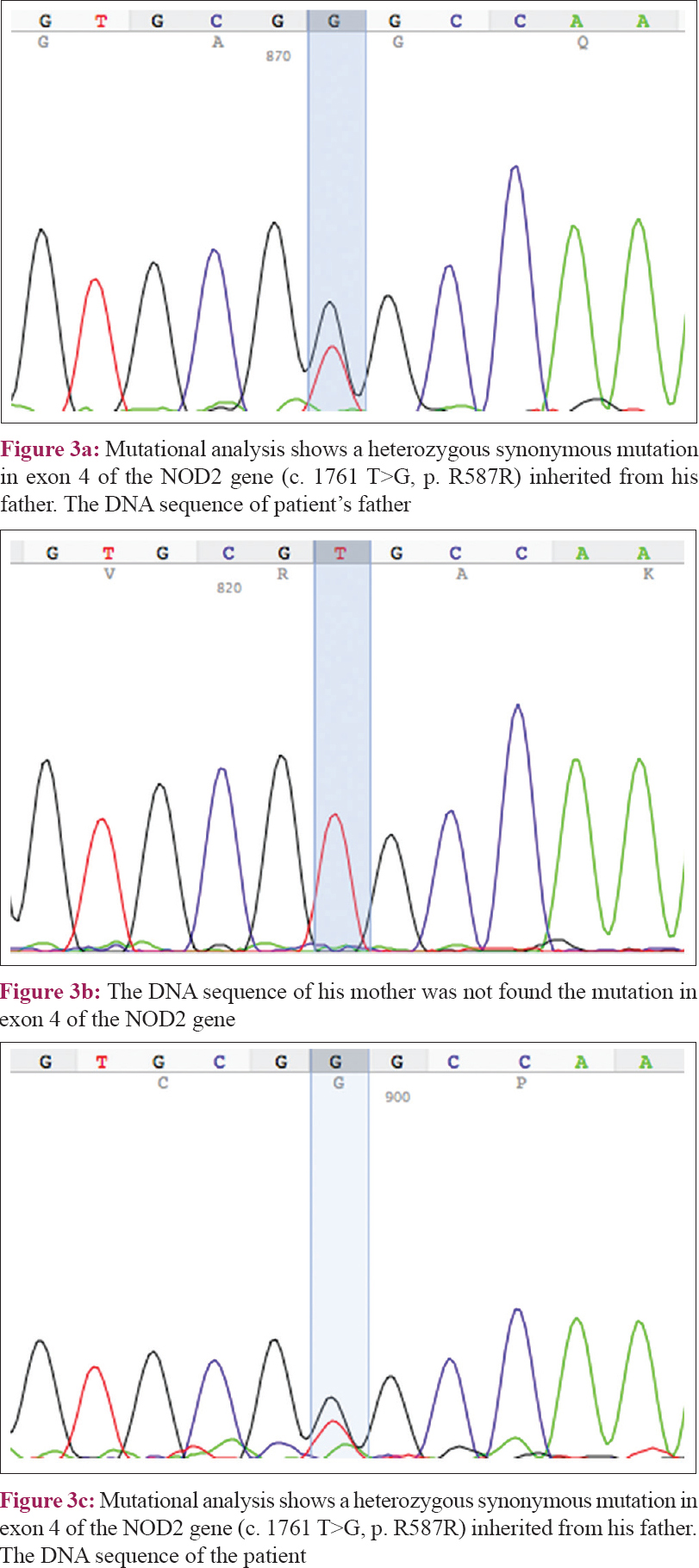 |
| Figure 3 |
The presence of clinical characteristics, typical histological hallmarks, along with a negative familial history supported a diagnosis of early-onset sarcoidosis, which was confirmed by the mutational analysis of the NOD2 gene. Treatment with oral prednisone (1 mg/kg/d) and MTX 5 mg/w for 2 months was associated with marked improvement.
Blau syndrome/early-onset sarcoidosis is mainly characterized by early onset and one or more recurrent manifestations of the triad of symptoms. Histological examination can confirm the presence of naked sarcoidal granulomas. The clinical picture needs to be corroborated by identification of the possible NOD2 gene mutation. The most frequently observed mutations of Blau syndrome/early-onset sarcoidosis are mis-sense substitutions involving exon 4, and the ones mainly affecting the arginine residue at position 334 (R334W/Q). The Y563H mutation in NOD2 has been described in five cases of 2 Brazilian families of Blau syndrome.[2] Our patient was born with congenital cleft lip and palate, which was not a feature reported in previous reports. Furthermore, we found a novel missense Y563D mutation in NOD2, which was not recognized in the genomes of his parents, confirming the sporadic origin of the mutation.
There is no established treatment modality for Blau syndrome/early-onset sarcoidosis. Nonsteroidal anti-inflammatory drugs, corticosteroids, and immunosuppressive drugs may be helpful. When patients are unresponsive to the combination of corticosteroids and immunosuppressant agents, the tumor necrosis factor-a inhibitor infliximab should be considered.[3] Our patient was given oral prednisone and methotrexate, resulting in dramatic improvement.
In conclusion, we report a sporadic clinical case of a Chinese patient suffering from early-onset sarcoidosis with congenital cleft lip and palate that reveal a novel missense mutation Y563D in the NOD2 gene.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Caso F, Galozzi P, Costa L, Sfriso P, Cantarini L, Punzi L. Autoinflammatory granulomatous diseases: From Blau syndrome and early-onset sarcoidosis to NOD2-mediated disease and Crohn's disease. RMD Open 2015;1:e000097.
[Google Scholar]
|
| 2. |
Jesus AA, Fujihira E, Watase M, Terreri MT, Hilario MO, Carneiro-Sampaio M, et al. Hereditary autoinflammatory syndromes: A Brazilian multicenter study. J Clin Immunol 2012;32:922-32.
[Google Scholar]
|
| 3. |
Caso F, Costa L, Rigante D, Vitale A, Cimaz R, Lucherini OM, et al. Caveats and truths in genetic, clinical, autoimmune and autoinflammatory issues in Blau syndrome and early onset sarcoidosis. Autoimmun Rev 2014;13:1220-9.
[Google Scholar]
|
Fulltext Views
2,403
PDF downloads
1,889





![[Figure - 1]](#fig_ijdvl_2018_84_5_645_217073_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2018_84_5_645_217073_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2018_84_5_645_217073_f3.jpg){kind=link}