
Translate this page into:
Identification of POLA1 gene deep intronic mutation confirms diagnosis of X-linked reticulate pigmentary disorder in a Chinese patient
Corresponding author: Dr. Aihua Wei, Beijing Tongren Hospital, Capital Medical University, 1 Dongjiaominxiang, Dongcheng District, Beijing, China. weiaihua3000@163.com and Dr. Xuyun Hu, Beijing Children’s Hospital, Capital Medical University, 56 South Lishi Road, Xicheng District, Beijing, China. hu_xuyun@126.com
-
Received: ,
Accepted: ,
How to cite this article: Zhang Y, Xie Y, Hu X, Wei A. Identification of POLA1 gene deep intronic mutation confirms diagnosis of X-linked reticulate pigmentary disorder in a Chinese patient. Indian J Dermatol Venereol Leprol. doi: 10.25259/IJDVL_623_2024
Dear Editor,
X-linked reticulate pigmentary disorder (XLPDR, OMIM#301220) is a rare disease characterised by diffuse reticulate dyschromatosis, distinctive facial features, ectodermal dysplasia, and immune dysregulation. In 2016, POLA1 was identified as the causative gene for XLPDR.1 POLA1 encodes the catalytic subunit of DNA polymerase-alpha, crucial for DNA replication.
A 24-year-old Chinese male presented with characteristic facial features and recurrent respiratory infections since six months of age, which improved after the age of 10. Hypopigmented macules appeared when he was six years old, and by 13 years of age, he began to experience vision loss, strabismus, and photophobia, which progressed to corneal scarring. He also had hypohidrosis affecting his head, face, and neck since birth with normal growth and mental development.
In 2015, the patient was clinically diagnosed with XLPDR after a dermatological examination that revealed generalised dark hyperpigmentation with reticular patterns and hypopigmented macules on the face and extremities [Figures 1a and 1b]. Routine laboratory tests, including blood, biochemical, and immunological tests, were normal. Ocular examination revealed corneal scarring, corneal edema on fundus photography and thickened corneal epithelial layers with scarring on optical coherence tomography [Figures 2a, 2b, and 2c]. Skin histopathology indicated hyperkeratosis of the epidermis and increased melanin granules in the keratinocytes of both the basal and spinous cell layers [Figure 3a]. Upon follow-up in 2023, there were no significant changes in skin or ocular symptoms, hypohidrosis, or immune profile.
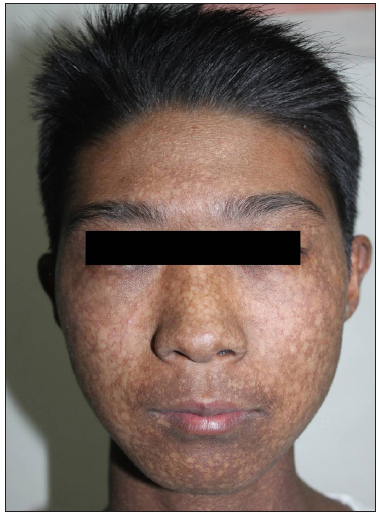
- Upswept hair, flared eyebrows, reticulate hyperpigmentation, and hypopigmentation on the face.
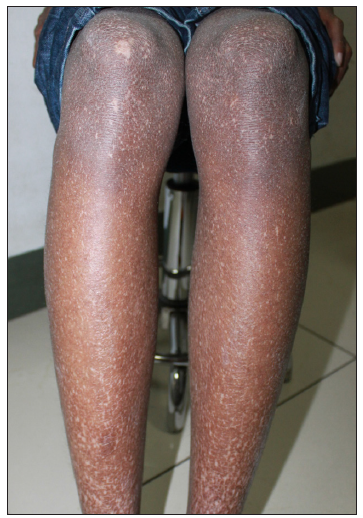
- Reticulate hyperpigmentation and hypopigmented patches on lower limbs.
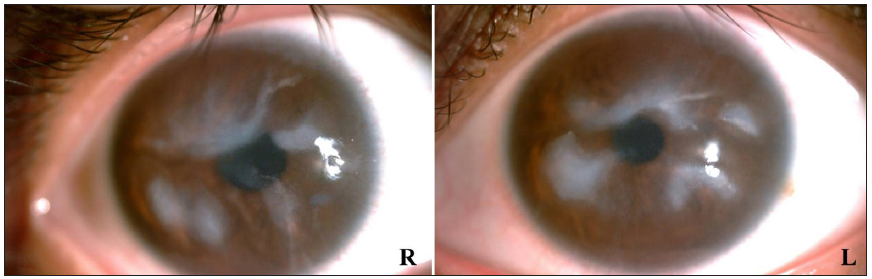
- Anterior segment examination at slit-lamp exhibiting corneal opacities.
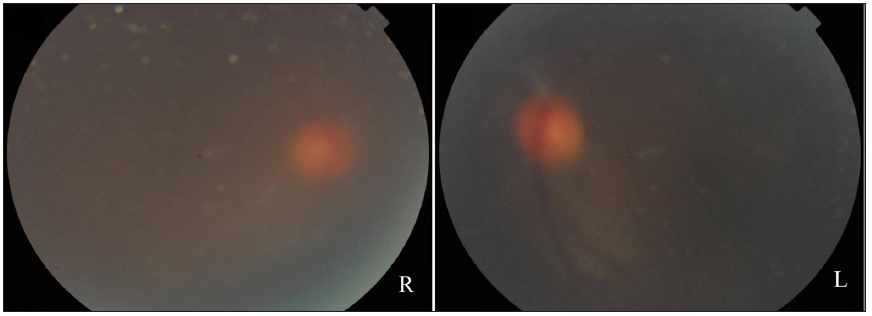
- Fundus photography showing a blurred fundus with a faintly visible normal optic disc.
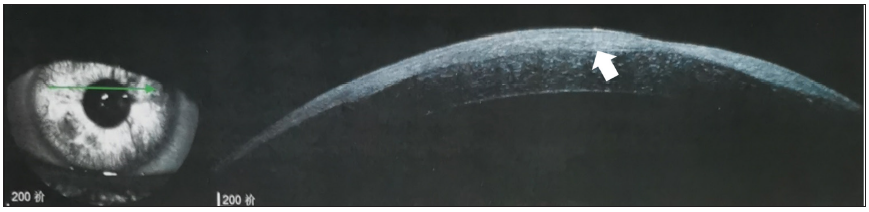
- Optical coherence tomography revealing oedematous thickening of the cornea (arrow), involving the corneal epithelial layer and scarring.
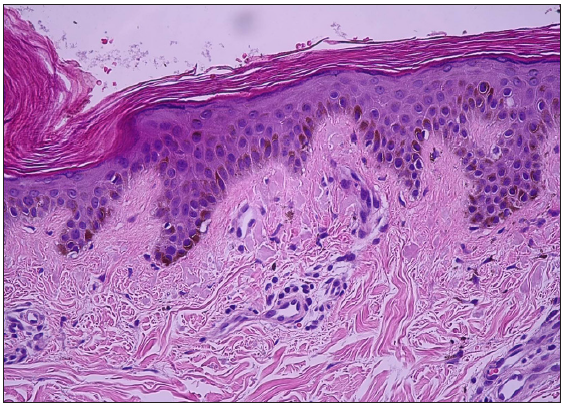
- Mild epidermolytic hyperkeratosis, basal layer hyperpigmentation, and scattered melanophages in the dermal papillae (Haematoxylin and eosin, 200x).
Whole-exome sequencing (WES) in 2015 did not identify any pathogenic variants. However, a follow-up reanalysis in 2023 using Sanger sequencing identified a previously reported deep intronic variant, c.1393-354A>G, in POLA1, which was inherited from the patient’s phenotypically normal mother [Figure 3b]. Notably, this deep intronic variant is classified as a pathogenic variant according to American College of Medical Genetics and Genomics (ACMG) guidelines, and it is the only known variant causing XLPDR.
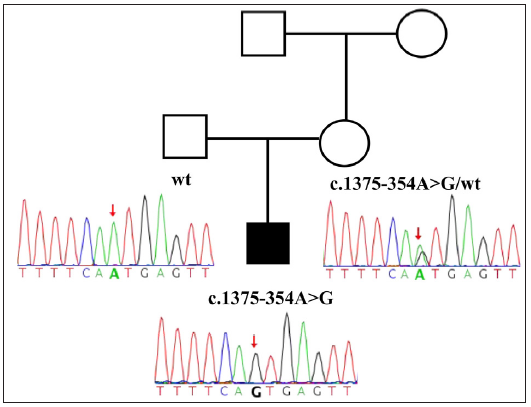
- Sanger sequencing results of the patient and his parents. The red arrow indicates the position of the mutation in the sequence. (R: right; L: left. A: Adenosine, C: Cytosine, T: Thymine, G: Guanine).
XLPDR is an exceptionally rare inherited pigmentary condition. Initially, Petro Starokadomskyy et al. identified the POLA1 gene variant c.1393-354A>G as the genetic cause of XLPDR in affected patients1. To date, approximately 30 cases have been reported but only seven of them had detailed clinical and genetic information documented. The clinical manifestations of these seven patients are summarised in Table 1. Functional experiments demonstrated that this deep intronic variant introduces a new exon, leading to a decrease in the expression level of the POLA1 protein, diminishing cytosolic RNA: DNA hybrids and further activating the transcription of the type I interferon gene. The type I interferon receptor amplifies signals through the JAK/STAT signalling pathway, triggering lymphocytes to produce excessive cytokines and inflammatory factors, resulting in a series of clinical symptoms.2
| Patient | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Reference | Starokadomskyy P et al. | Xu Z et al. | Légeret C et al. | Zhao YK et al. | Li XY et al. | Li MWY et al. | Present case |
| Origin | American | Chinese | Caucasian | Chinese | Chinese | Vietnamese | Chinese |
| Year of publication | 2017 | 2019 | 2021 | 2022 | 2023 | 2024 | N/A |
| Pubmed ID | 28407217 | 30714101 | 32989594 | 35645674 | 37851432 | 38165470 | N/A |
| Variant | c.1393-354A>G | c.1393-354A>G | c.1393-354A>G | c.1393-354A>G | c.1393-354A>G | c.1393-354A>G | c.1393-354A>G |
| Method of identification | N/A | N/A | Targeted sequencing | Exome sequencing | Genome sequencing | Genome sequencing | Sanger sequencing |
| Sex | Male | Male | Male | Male | Male | Male | Male |
| Age | 13y | 26y | 12y | 4y | 7y | 9y | 24y |
| Corneal scar | N/A | N/A | N/A | N/A | N/A | Yes | Yes |
| Photophobia | Yes (7y) | N/A | N/A | Yes (3m) | Yes | Yes | Yes |
| Skin Hyperpigmentation | Yes (1y) | Yes (early childhood) | Yes | Yes (6m) | Yes (1y) | Yes (8m) | Yes |
| Hypohidrosis | Yes | Yes (early childhood) | N/A | Yes | Yes | Yes | Yes |
| Upswept frontal hairline | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Flared eyebrows | Yes | Yes | Yes | Yes | N/A | Yes | Yes |
| Respiratory System Infections | Yes (1y) | Yes (childhood) | Yes (18m) | No | Yes | Yes | Yes |
| Gastrointestinal Issues | Yes (1y) | N/A | Yes (18m) | N/A | Yes | Yes | No |
| Urogenital involvement | Yes (1y) | N/A | No | N/A | N/A | No | No |
N/A: Not available; y: year; m: month; XLPDR: X-linked reticulate pigmentary disorder.
The onset time of the symptoms is indicated in parentheses.
Pigmentary abnormalities in XLPDR typically appear between the ages of four months and five years.3 However, our patient did not exhibit skin lesions in infancy. Hypopigmented spots emerged at six years old, progressing to reticular hyperpigmentation, which is an atypical presentation not previously reported. Throughout the two visits, the patient’s generalised skin reticular pigment abnormality showed no significant worsening over an eight-year period. Currently, all patients with XLPDR have skin pigmentation. The POLA1 gene is mainly involved in DNA replication and is significantly expressed in skin melanocytes. However, there are few studies on how the POLA1 gene affects the function of melanocytes and the mechanism of melanin production, and further research is needed to clarify this.
Initially, whole-exome sequencing in 2015 failed to identify a causative variant for our patient, relying solely on clinical assessment for diagnosis. Subsequent Sanger sequencing, performed several years later, confirmed the presence of the POLA1 variant c.1393-354A>G, which had been previously reported in other XLPDR patients. Direct detection of this variant proves to be a more effective and cost-efficient approach for individuals with XLPDR. Our experience underscores the importance of reanalysis for cases with initially negative results in genetic diagnosis. Currently, over half of patients with rare genetic disease remain undiagnosed after WES testing, partly due to the limited scope of the targeted regions in exome sequencing.4 Similarly, the pathogenic deep intronic variant in POLA1 was identified only through whole-genome sequencing (WGS) years after clinical diagnosis.1 Furthermore, with the increasing number of individuals undergoing genetic testing, approximately 300 or more new gene–disease associations are discovered annually.5 Therefore, reanalysis of patients with negative genetic diagnoses every two to five years has a high potential for uncovering new genetic etiologies.6,7
Currently, there is no effective treatment for XLPDR. However, our patient exhibited well-controlled lesions with no evident progression and were observed with strict sun protection. Prompt consultation with a doctor is essential for any eye symptoms to prevent irreversible corneal scarring due to recurrent keratitis. Basic supportive treatment is essential for managing multisystemic involvements, and it is noted that multisystemic infections may naturally improve with age.
In conclusion, our study underscores the importance of reanalysis and the role of deep intronic variants in rare genetic disorders like XLPDR, contributing valuable insights to the growing understanding of how such genetic variations can be overlooked in initial diagnostic attempts.
Ethical approval
The research/study was approved by the Institutional Review Board No.: TREC2023-KYS088; dated 2023.02.24 at the Ethics Committee of Beijing Tongren Hospital, Capital Medical University.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
This work was partially supported by Beijing Natural Science Foundation (7234355).
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- DNA polymerase-α regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nat Immunol. 2016;17:495-504.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Immune dysfunction in mendelian disorders of POLA1 deficiency. J Clin Immunol. 2021;41:285-93.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- X-linked reticulate pigmentary disorder with systemic manifestations: A new family and review of the literature. Am J Med Genet A. 2013;161A:1414-20.
- [CrossRef] [PubMed] [Google Scholar]
- Increasing the sensitivity of clinical exome sequencing through improved filtration strategy. Genet Med. 2017;19:593-8.
- [CrossRef] [PubMed] [Google Scholar]
- International cooperation to enable the diagnosis of all rare genetic diseases. Am J Hum Genet. 2017;100:695-705.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Evaluating systematic reanalysis of clinical genomic data in rare disease from single center experience and literature review. Mol Genet Genomic Med. 2020;8:e1508.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Re-evaluation and re-analysis of 152 research exomes five years after the initial report reveals clinically relevant changes in 18. Eur J Hum Genet. 2023;31:1154-64.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




