Translate this page into:
Immunofluorescence antigen mapping for hereditary epidermolysis bullosa
2 Department of Dermatology, St. John's Institute of Dermatology, St. Thomas' Hospital, London, SE1 7EH, United Kingdom,
Correspondence Address:
Raghavendra Rao
Department of Dermatology, Kasturba Medical College, Manipal University, Manipal, Karnataka, India
| How to cite this article: Rao R, Mellerio J, Bhogal BS, Groves R. Immunofluorescence antigen mapping for hereditary epidermolysis bullosa. Indian J Dermatol Venereol Leprol 2012;78:692-697 |
Abstract
Epidermolysis bullosa (EB) is a group of inherited, mechanobullous disorders that are caused by mutations in the structural proteins in the epidermis or dermoepidermal junction. Characteristic clinical picture is the presence of blisters at trauma prone areas of the body, which develops at or soon after birth. Availability of specific monoclonal antibodies against the target proteins together with advances in the molecular genetics have led to the revision in the classification of EB. Now four major types of EB are recognized depending upon the level of blister and the location of target protein: EB simplex (epidermolytic), junctional EB (lucidolytic), dystrophic EB (dermolytic) and Kindler's syndrome (mixed cleavage plane). The laboratory tests not only help to confirm the diagnosis of EB but are also an important tool to classify (and subtype) EB. These include immunofluorescence antigen mapping (IFM), transmission electron microscopy (TEM) and mutation analysis. IFM is the most preferred method for final diagnosis of EB worldwide. It is relatively easy to perform and results can be obtained rapidly. This article describes the technicalities and significance of IFM in various types of EB.Epidermolysis bullosa (EB) is a group of genetic blistering diseases that is characterised by fragility of the skin and mucous membranes. [1] Blisters and erosions which appear at birth or shortly thereafter are typically seen on the trauma prone areas of the body. EB is characterized by extensive phenotypic variability with considerable morbidity and mortality. In the milder forms there is a lifelong blistering tendency with no impact on the overall longevity of the affected individual, while in the most severe forms children die during the early postnatal period from metabolic perturbations, dehydration, and sepsis. [2] The principal forms of hereditary EB are the consequence of mutations in genes coding for proteins involved in epidermal keratinocyte-basement membrane zone (BMZ) adhesion [Figure - 1]. To date, more than 1000 mutations in at least 14 structural genes have been documented resulting in defective adhesion, clinically manifesting as blister formation. [3],[4],[5]
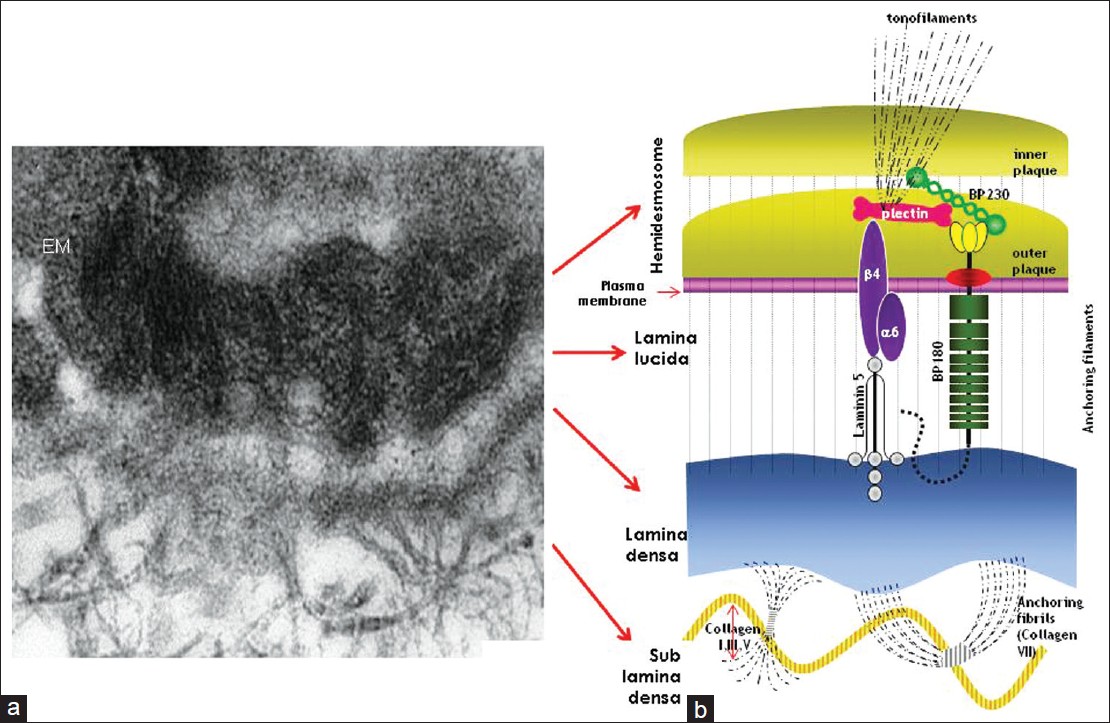 |
| Figure 1: Electron micrograph of cutaneous basement membrane zone (a) and schematic illustration of molecular components which are involved in different form of EB (b) Courtesy: St. John's Institute of Dermatology, London |
Classification of Epidermolysis Bullosa
Pearson in 1962 proposed a sophisticated classification system for EB based on the findings of transmission electron microscopy (TEM). [6] Depending on the ultrastructural levels within which the split develops in EB skin, either spontaneously or following minor trauma, he classified EB into three major types: epidermolytic (EB simplex), lucidolytic (junctional EB) and dermolytic (dystrophic EB). In EB simplex (EBS), blistering develops within the epidermis; in junctional EB (JEB), the level of cleavage is within lamina lucida while in dystrophic EB (DEB) blister formation occurs in the sublamina densa zone. In 2007, the third international consensus meeting on diagnosis and classification of EB was held in Vienna, Austria. [7] Based on the outcome of this meeting, EB is now classified into 4 major types; mixed type (Kindler syndrome) being the fourth major type [Table - 1]. Availability of monoclonal and polyclonal antibodies coupled with advances in molecular diagnostic techniques have led to the subclassification of EB into at least 30 different subtypes. [8] However, the classification of EB is still evolving; since the third consensus classification was published, a new but rare subtype of recessive EBS resulting from mutations in the dystonin gene (which is homologous to BP230 antigen) has been described, characterized phenotypically by localized blistering and neurological impairment. [9]
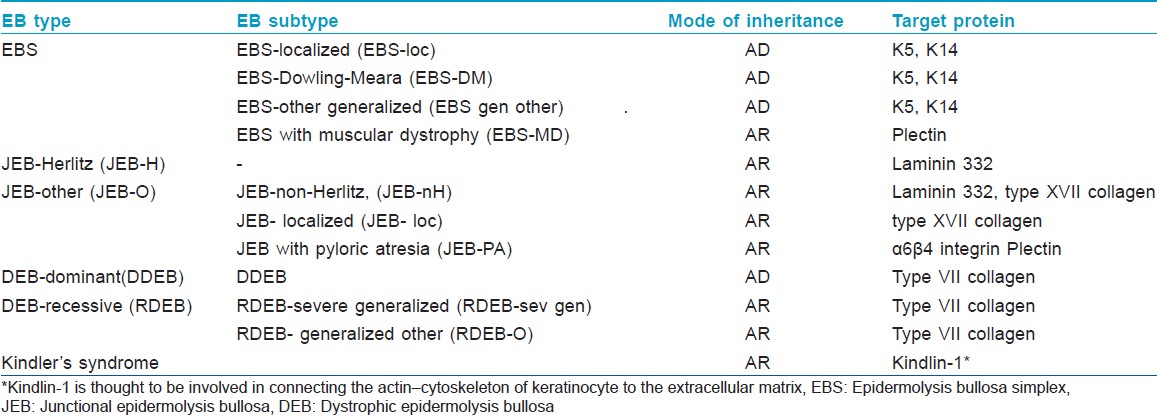
Need For Laboratory Diagnosis of Epidermolysis Bullosa
The severity and natural course of EB depends on the nature of the affected protein and the causative mutation(s). Major types of EB may be clinically indistinguishable, especially in the neonatal period. [10] Furthermore, there is significant phenotypic overlap between different types of EB. A classification based solely on clinical grounds may be inaccurate. For example, clinical signs such as atrophic scarring, milia formation and nail dystrophy can occur in each of the 4 major EB subtypes. Further, scarring and nail changes may not be evident in infancy even in severe forms of EB such as JEB and DEB suggesting an erroneous diagnosis of EBS. For these reasons, determination of major EB type should be confirmed with immunofluorescence mapping (IFM) or TEM to enable informed discussion regarding prognosis. [7]
Laboratory Diagnosis of Epidermolysis Bullosa
It is important to stress that routine histological examination of skin is not recommended in EB, since it may be difficult or impossible to distinguish at the light microscopy level between even lower intra-epidermal and subepidermal cleavage. Similarly, the precise distinction between intra-lamina lucida (i.e., JEB) and sublamina densa (i.e., DEB) cleavage cannot be ascertained by light microscopy. [8] More advanced diagnostic techniques such as IFM or TEM should be employed; they not only help us to diagnose JEB and DEB with a great degree of certainty but also form the basis for further molecular testing by mutation analysis. [11] Information obtained by these tests will enable the clinician to counsel patients and parents about the prognosis of the disease.
TEM is considered the gold standard laboratory test for differentiation between the various forms of EB. The primary advantage of TEM is that it can visualize ultra-structural abnormalities and provide a semi-quantitative assessment of specific BMZ structural deficits. [12] It may be particularly useful in patients with mild DEB or EBS; IFM may be normal in these cases but TEM shows morphological abnormalities of anchoring fibrils or intermediate filaments (or clumped tonofilaments in the case of EBS-Dowling Meara), respectively. Sometimes a split may not be visible in an IFM sample, but TEM can show an ultrastructural split. Multiple cleavage planes as seen in Kindler syndrome may be appreciated only by TEM. However, TEM is time-consuming and expensive, and the results are, to a high degree, operator-dependent and sometimes inaccurate. In addition, there are only a few laboratories in the world today with appropriate experience and skills to analyze and interpret EB samples by TEM. [11] The diagnostic precision of IFM is similar to that of TEM with the advantage that it is simpler and faster both to perform and to interpret. [13] A previous study has shown that IFM is more sensitive (97% vs. 71%) and specific (100% vs. 81%) than TEM. [14] IFM has other distinct advantages. Firstly, it is less expensive than TEM. Secondly, with the use of specific monoclonal antibodies, it can provide considerable insight into not only the major subtypes of EB but also to the most likely mutated structural protein. [7] Finally, a biopsy sample for IFM can be stored in Michel′s transport medium at room temperature for up to 28 days; hence, can be transported worldwide for evaluation. [15] A recent study has also shown the utility of IFM in the prenatal diagnosis of certain types of severe EB by studying first trimester chorionic villous biopsy. [16]
Immunofluorescence Mapping
In 1981, Hintner et al described the technique of IFM; using antibodies against BMZ components, they described different staining patterns in the major types of EB. [13] IFM could be considered as an indirect immunofluorescence technique, since it is first necessary to promote formation of an immune-complex by adding a primary antibody to the tissue under investigation. [5] The primary antibodies are mostly monoclonal belonging to IgG class and are raised in mouse against human proteins. Then, a secondary antibody tagged with fluorochrome is used to reveal this immune-complex. The secondary antibodies are usually anti-IgG mouse specific antibodies; an exception is the antibody against α6 integrin, where rat is the source of primary antibody, and thus the secondary antibody has to be anti-rat IgG. [11] Fluorochromes are dyes that emit light at a specific wavelength when stimulated by ultraviolet radiation. The most commonly used fluorochrome is fluorescein, which is lime-green in color. Depending on the antibody used, this technique confirms whether the expression of proteins is normal, reduced or absent [Figure - 2]. Currently, several antibodies [Table - 2] that recognize the pathological proteins in keratinocytes and BMZ are available commercially. [17]
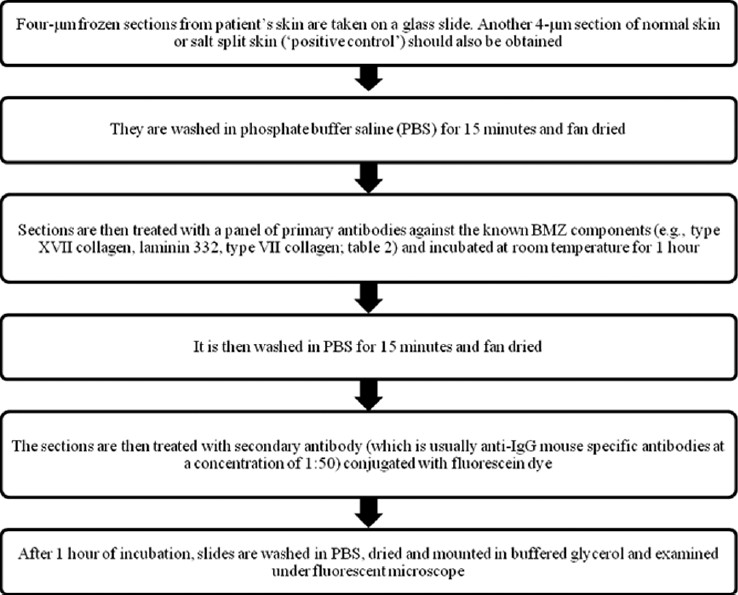 |
| Figure 2: Immunofluorescence mapping technique |

Biopsy Technique
The most critical step of IFM is taking the skin biopsy correctly and transporting it to the laboratory. [4] It is ideal to take biopsy from an artificially induced blister. A blister may be induced by gently rubbing the skin to produce mild erythema. An advantage of this method is that this will contain a cleavage plane without secondary changes. Rubbing is performed by applying firm downward pressure with an eraser or finger and then rotating it laterally (at least 180 degrees each way). This should be continued for at least 20 times or till the area turns red. One should ideally wait for 5 minutes for a blister to develop microscopically before taking the biopsy. In cases where the skin appears extremely fragile (e.g., severe JEB and DEB) it may be impossible to pre-rub the skin since this leads to macroscopic blistering and subsequent total epidermal separation when the biopsy is taken. The cleavage planes may develop in the skin in these conditions just with a routine punch biopsy technique. [18] Although, biopsies are sometimes taken from fresh blisters, ideally this should be avoided because it may give false positive results due to proteolytic antigen degradation or re-epithelialization under the roof of the blister, resulting in multiple cleavage planes. [19] When a blister biopsy is contemplated, another a 3-mm punch biopsy from an unaffected non-rubbed area, usually the inner upper arm should be taken as the reduction of protein staining, if present, is more easily assessed on skin that has not actually blistered.
A 3-mm punch or shave biopsy should be taken after proper antiseptic precautions and anaesthetization. Shave biopsy is the preferred method at the St. John′s Institute as the rate of cleavage has been found to be unacceptably high with punch biopsy. The biopsy specimen is then placed in Michel′s medium and transported to laboratory. If two biopsies are contemplated then they should be sent to the laboratory in different vials after proper labeling. Another minor but important issue is that when biopsy is planned for routine histopathological section (hematoxylin and eosin) along with IFM, the one for immunofluorescence study should always be done first as there may be a possibility of formalin contamination which renders biopsy specimen suboptimal for IFM studies.
Steps of Immunofluorescence Mapping
Interpretation of results
Epidermolysis bullosa simplex
In cases of EBS, all antibodies are found at the base of the blister. Additional EBS-specific antibodies (keratin 5 and 14, plectin and α6β4 integrin) may be employed; in general, expression of proteins are normal, except in autosomal recessive EBS, when patients may have absent keratin 14 staining. [20] In patients with EBS-muscular dystrophy (EBS-MD), plectin is mostly absent, and in the rare case of EBS Ogna, it is markedly reduced. [21] In EB subtypes with pyloric atresia (either EBS or JEB), plectin or, α6β4 integrin are reduced or absent. [22] Collagen IV staining, which highlights the lamina densa, is along the base of the blister.
Junctional epidermolysis bullosa
The main target proteins in JEB are type XVII collagen (BP180 or BPAG2) and laminin 332 (previously laminin 5). Collagen XVII is expressed on the roof of split skin whereas other antibodies are seen on the floor of the blister. In the severe Herlitz form of JEB (JEB-H), caused by mutations in one of the genes encoding the three polypeptide chains of laminin 332, expression of this protein is absent or markedly reduced [Figure - 3]. In cases of non-Herlitz JEB (JEB-nH) there is reduced staining of laminin 332. In cases where there is a collagen XVII mutation, there is marked reduction or absence of expression of collagen at the BMZ with normal expression of laminin 332. In JEB with pyloric atresia, staining to α6 and β4 integrin subunits is reduced or absent. [11] Type IV collagen staining in all forms of JEB localizes to the blister floor.
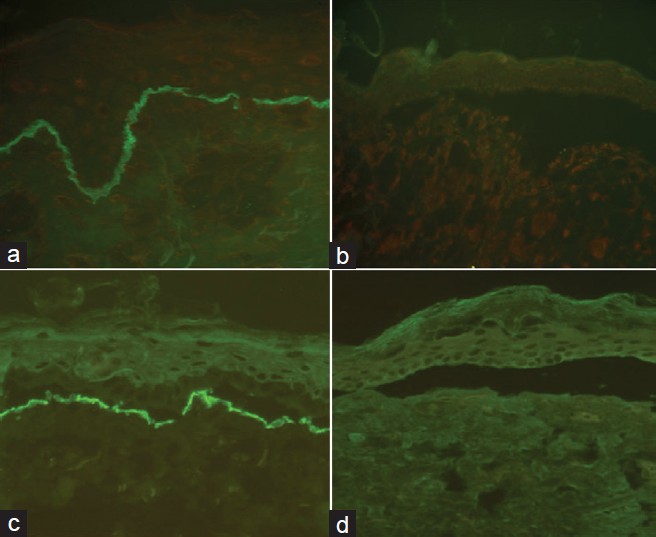 |
| Figure 3: Photomicrograph showing normal expression of laminin 332 in the control skin and complete absence of staining in JEB-H (a and b, respectively) and normal expression of type VII in the control skin and complete absence of staining in RDEB (c and d, respectively) Courtesy: St. John's Institute of Dermatology, London |
Dystrophic epidermolysis bullosa
All DEB subtypes are caused by mutations in type VII collagen which is the principal component of anchoring fibrils. The level of cleavage occurs in the sublamina densa with collagen XVII and laminin 332 staining seen in the roof of the blister. In patients with severe generalized recessive DEB (RDEB), IFM shows absent or barely detectable type VII collagen [Figure - 3]. In these cases, immunostaining of type IV collagen occurs on the roof and indicates dermolytic blistering to confirm DEB. Other generalized or localized subtypes of DEB may show a reduced or normal expression of type VII collagen. [11],[17]
Kindler′s syndrome
IFM using a standard panel of BMZ antibodies does not show any reduction or major alteration in staining intensity though type IV and VII collagen antibodies may show broad, reticular staining at the dermal-epidermal junction. Labeling of normal skin with a novel, polyclonal antibody against kindlin-1 shows a bright staining in the epidermis, particularly in the basal keratinocytes and along the dermal-epidermal junction without any dermal alterations. In contrast, in Kindler syndrome skin there is a marked reduction and, in some cases, a complete absence of staining in the epidermis. [23]
Conclusions
IFM has taken over from TEM as the preferred method for preliminary diagnosis of EB. This is due to various reasons including its cheaper cost, easy shipping of samples, speedy results and less reliance on highly specialized expertise to carry it out and interpret results. The use of various antibodies and observation of staining patterns has not only enabled subclassification of EB more precisely but also allows identification of candidate proteins as well as blister cleavage planes.
| 1. |
Uitto J, Richard G. Progress in epidermolysis bullosa: From eponyms to molecular genetics classification. Clin Dermatol 2005;23:33-40.
[Google Scholar]
|
| 2. |
Varki R, Sadowski S, Pfendner E, Uitto J. Epidermolysis bullosa. I. Molecular genetics of the junctional and hemidesmosomal variants. J Med Genet 2006;43:641-52.
[Google Scholar]
|
| 3. |
Shinkuma S, McMillan JR, Shimizu H. Ultrastructure and molecular pathogenesis of epidermolysis bullosa . Clin Dermatol 2011;29:412-9.
[Google Scholar]
|
| 4. |
Intong LRA, Murrell DF. Inherited epidermolysis bullosa: New diagnostic criteria and classification. Clin Dermatol 2012;30:70- 7.
[Google Scholar]
|
| 5. |
Oliveira ZN, Périgo AM, Fukumori LM, Aoki V. Immunological mapping in hereditary epidermolysis bullosa. An Bras Dermatol 2010;85:856-61.
[Google Scholar]
|
| 6. |
Pearson RW. Studies on the pathogenesis of epidermolysis bullosa. J Invest Dermatol 1962;39:551-75.
[Google Scholar]
|
| 7. |
Fine JD, Eady RA, Bauer EA, Bauer JW, Bruckner-Tuderman L, Heagerty A, et al. The classification of inherited epidermolysis bullosa (EB): Report of the Third International Consensus meeting on Diagnosis and Classification of EB. J Am Acad Dermatol 2008;58:931-50.
[Google Scholar]
|
| 8. |
Fine JD. Inherited epidermolysis bullosa. Orphanet J Rare Dis 2010;5:12.
[Google Scholar]
|
| 9. |
Groves RW, Liu L, Dopping-Hepenstal PJ, Markus HS, Lovell PA, Ozoemena L, et al. A homozygous nonsense mutation within the dystonin gene coding for the coiled-coil domain of the epithelial isoform of BPAG1 underlies a new subtype of autosomal recessive epidermolysis bullosa simplex. J Invest Dermatol 2010;130:1551-7.
[Google Scholar]
|
| 10. |
Nischler E, Klausegger A, Hüttner C, Pohla-Gubo G, Diem A, Bauer JW, et al. Diagnostic pitfalls in newborns and babies with blisters and erosions. Dermatol Res Pract. Available from: http://www.hindawi.com/journals/drp/2009/320403/ [Last accessed on 2011 Dec 25].
[Google Scholar]
|
| 11. |
Pohla-Gubo G, Cepeda-Valdes R, Hintner H. Immunofluorescence Mapping for Epidermolysis Bullosa. Dermatol Clin 2010;28:201- 10.
[Google Scholar]
|
| 12. |
McMillan JR, McGrath JA, Tidman MJ, Eady RA. Hemidesmosomes show abnormal association with the keratin filament network in junctional forms of epidermolysis bullosa. J Invest Dermatol 1998;110:132-7.
[Google Scholar]
|
| 13. |
Hintner H, Stingl G, Schuler G, Fritsch P, Stanley J, Katz S, et al. Immunofluorescence mapping of antigenic determinants within the dermal-epidermal junction in the mechanobullous diseases. J Invest Dermatol 1981;76:113-8.
[Google Scholar]
|
| 14. |
Yiasemides E, Walton J, Marr P, Villanueva EV, Murrell DF. A comparative study between transmission electron microscopy and immunofluorescence mapping in the diagnosis of epidermolysis bullosa. Am J Dermatopathol 2006;28:387-94.
[Google Scholar]
|
| 15. |
Vaughan Jones SA, Bhogal BS, Black MM. The use of Michel's transport media for immunofluorescence and immunoelectron microscopy in autoimmune bullous diseases. J Cutan Pathol 1995;22:365-70.
[Google Scholar]
|
| 16. |
D'Alessio M, Zambruno G, Charlesworth A, Lacour JP, Meneguzzi G. Immunofluorescence analysis of villous trophoblasts: A tool for prenatal diagnosis of inherited epidermolysis bullosa with pyloric atresia. J Invest Dermatol 2008;128:2815-9.
[Google Scholar]
|
| 17. |
Cepeda-Valdés R, Pohla-Gubo G, Borbolla-Escoboza JR, Barboza-Quintana O, Ancer-Rodríguez J, Hintner H, et al. Immunofluorescence mapping for diagnosis of congenital epidermolysis bullosa. Actas Dermosifiliogr 2010;101:673-82.
[Google Scholar]
|
| 18. |
Intong LRA, Murrell DF. How to take skin biopsies for epidermolysis bullosa. Dermatol Clin 2010;28:197-200.
[Google Scholar]
|
| 19. |
Fine JD, Burge SM. Genetic blistering diseases. In: Burns DA, Breathnach S, Cox N, Griffiths C, editors. Rook's textbook of dermatology. 7 th ed. Oxford: Wiley Blackwell; 2010. p. 39.1-39.37.
th ed. Oxford: Wiley Blackwell; 2010. p. 39.1-39.37. '>[Google Scholar]
|
| 20. |
Yiasemides E, Trisnowati N, Su J, Dang N, Klingberg S, Marr P, et al. Clinical heterogeneity in recessive epidermolysis bullosa due to mutations in the keratin 14 gene, KRT14. Clin Exp Dermatol 2008;33:689-97.
[Google Scholar]
|
| 21. |
Koss-Harnes D, Hoyheim B, Anton-Lamprecht I, Gjesti A, Jorgensen RS, Jahnsen FL, et al. A site-specific plectin mutation causes dominant epidermolysis bullosa simplex Ogna: Two identical de novo mutations. J Invest Dermatol 2002;118:87-93.
[Google Scholar]
|
| 22. |
Pfendner EG, Lucky AW. Epidermolysis Bullosa with Pyloric Atresia. In: Pagon RA, Bird TD, Dolan CR, Stephens K, editors. Gene Reviews. Seattle (WA): University of Washington, Seattle; 1993-2008 Feb 22 [Last updated on 2009 Apr 28]. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20301336 [Last accessed on 2011 Dec 27].
[Google Scholar]
|
| 23. |
Ashton GH. Kindler's syndrome. Clin Exp Dermatol 2004;29:116-21.
[Google Scholar]
|
Fulltext Views
7,218
PDF downloads
2,227





![[Figure - 1]](#fig_ijdvl_2012_78_6_692_102358_f3.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2012_78_6_692_102358_t1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2012_78_6_692_102358_f4.jpg){kind=link}
![[Table - 2]](#tbl_ijdvl_2012_78_6_692_102358_t2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2012_78_6_692_102358_f5.jpg){kind=link}