
Translate this page into:
Kikuchi-Fujimoto disease and Behcet’s disease: A rare association

Corresponding author: Dr. Suman Patra, Department of Dermatology, All India Institute of Medical Sciences, Jodhpur, Rajasthan, India. patrohere@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Bhatia D, Patra S, Verma N, Elhence PA. Kikuchi-Fujimoto disease and Behcet’s disease: A rare association. Indian J Dermatol Venereol Leprol. doi: 10.25259/IJDVL_196_2025
Dear Editor,
Kikuchi-Fujimoto disease is a rare disorder characterised by painful necrotising regional lymphadenopathy that warrants the evaluation of associated systemic diseases like autoimmune diseases, vasculitis, and infections.1,2 Hereby, we report a unique case of Kikuchi-Fujimoto disease in a patient with Behçet disease.
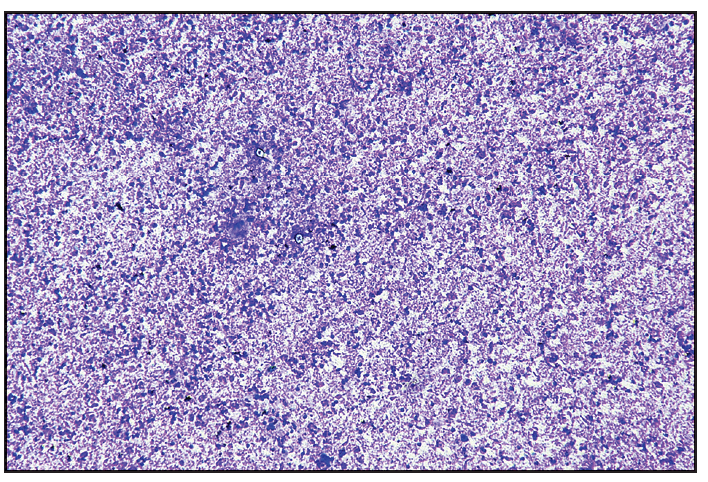
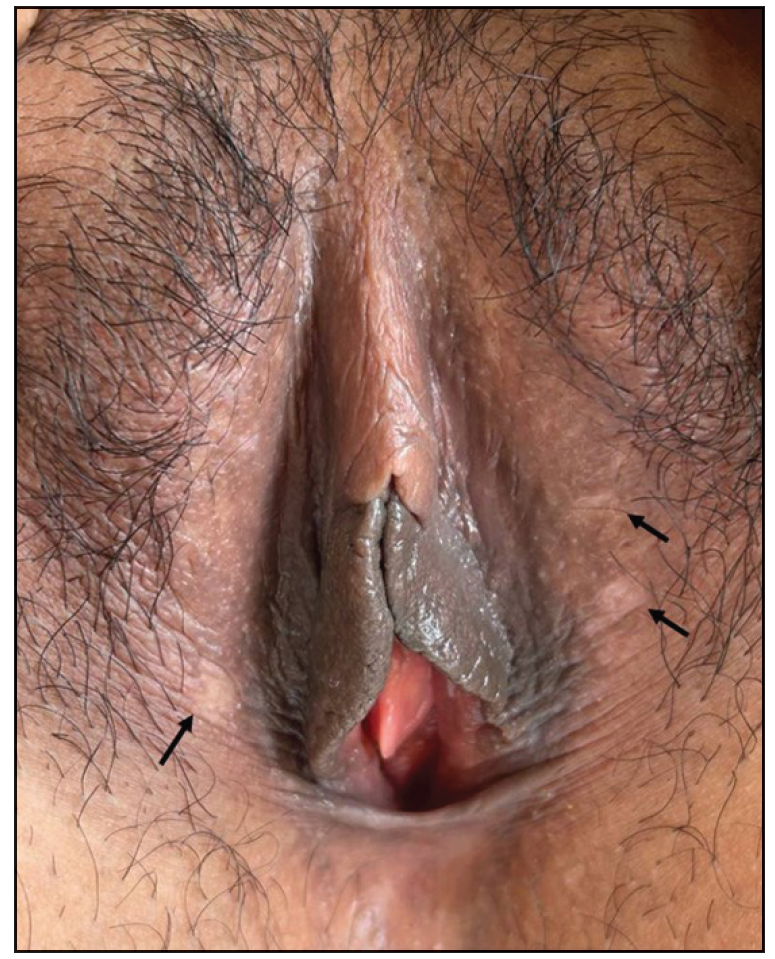
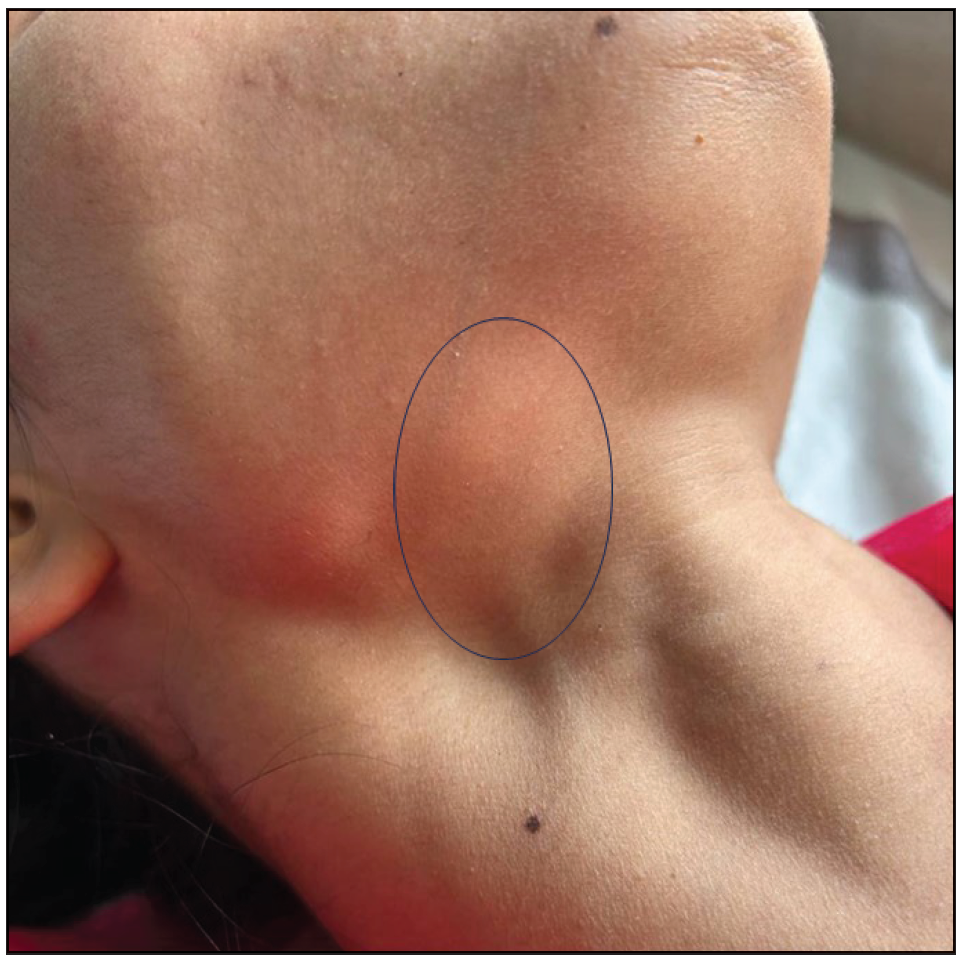
A 26-year-old woman presented with recurrent oral and genital ulcers for 8 months associated with blurred vision and pain in multiple large joints. She developed multiple painful swellings on the neck and axilla, with a low-grade fever. Examination revealed multiple aphthous ulcers up to 0.5 cm in size on the vulva and atrophic scars on the labia majora, indicating a healed lesion [Figure 1a]. There were tender, non-matted cervical and axillary lymph nodes. [Figures 1b]. Ophthalmological evaluation revealed scleritis in her left eye. There was no history suggestive of connective tissue disorders, productive cough, loss of appetite, significant weight loss, headaches, or seizure. A diagnosis of Behcet disease was considered and investigated. Her Human leukocyte antigen (HLA)-B51 analysis and pathergy test were positive. Her complete blood count, liver and kidney function tests, urine microscopy, Antinuclear antibodies, Chest x-ray, and mantoux test were normal. The International Study Group criteria revealed a score of 7 classifying as Behcet disease. Fine needle aspiration cytology (FNAC) of the affected lymph node showed polymorphous small mature lymphocytes, plasma cells, and few neutrophils and eosinophils along with lympho-histiocytic tangles and capillary strands in a necrotic background with karyorrhectic and calcified debris suggestive of necrotising lymphadenitis [Figure 2a-c]. No acid-fast bacilli were seen with the modified Ziehl-Neelsen stain, and CBNAAT (Cartridge-Based Nucleic Acid Amplification Test) was negative. A diagnosis of Kikuchi-Fujimoto disease coexistent with Behcet disease was made. She was treated with oral colchicine 0.5 mg two times a day, after which she had complete resolution of ulcers and significant improvement in lymphadenopathy on four weeks of follow-up [Figures 3a and 3b].
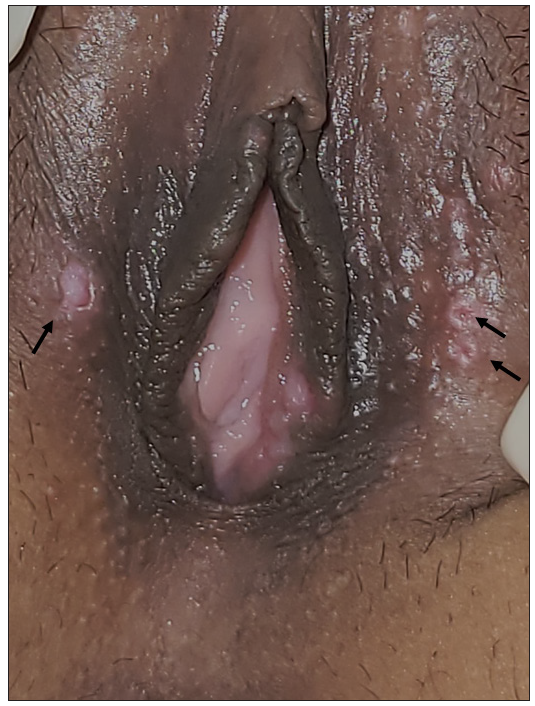
- Multiple well-defined aphthous ulcers on the labia majora as shown by black arrows.
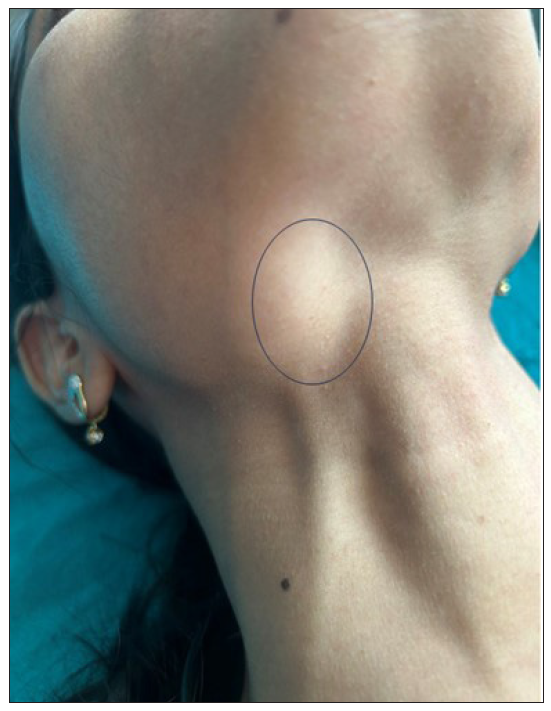
- Lymphadenopathy became more prominent on deglutition as marked by a black circle.
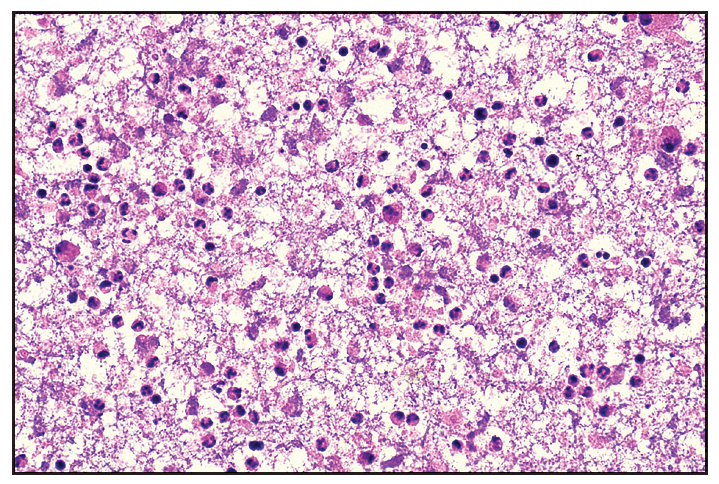
- Fine needle aspiration cytology (FNAC) of affected lymph node shows moderate cellularity with acute on chronic inflammatory infiltrate comprising of polymorphous population of small mature lymphocytes, plasma cells, few neutrophils and eosinophils along with lympho-histiocytic tangles and capillary strands in a necrotic background. Karyorrhectic and calcific debris and RBCs seen (Haematoxylin and eosin, 40x).
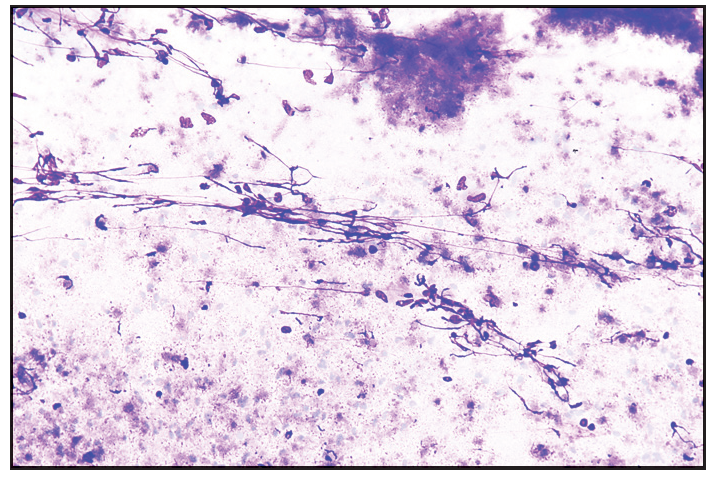
- Moderate cellularity with acute on chronic inflammation in a necrotic background. Few histiocytes and necrotic granular material also seen (Giemsa, 20x).
- Necrotic granular material along with inflammation (Giemsa, 10x)
- Complete healing of genital aphthous ulcers with mild atrophy and hypopigmentation as shown by black arrows.
- Decrease in the size of the right submandibular cervical lymph node as shown in a black circle.
Kikuchi-Fujimoto disease is more prevalent in young adults of Asian descent and is characterised by necrotising cervical lymphadenopathy with low-grade fever.2 It is usually associated with systemic disorders, which include systemic lupus erythematosus, antiphospholipid syndrome, thyroiditis, polymyositis, scleroderma, and Still’s disease. Behcet disease is multifactorial, and various infectious, genetic, and immunological factors contribute to the pathogenesis.3 There is limited literature available on the association between these two disorders. A study of 102 patients with Kikuchi-Fujimoto disease found two with coexistent Behcet disease. Both had recurrent Kikuchi-Fujimoto disease, which appeared after the onset of Behcet disease.4 Another report mentioned Kikuchi-Fujimoto disease in a patient with neuro-Behcet disease.5 It has been postulated that a few apoptotic cells arise from necrotising lymphadenopathy and autoantigens in various autoimmune disorders.6 Activation of the immune system by various triggers, leading to the production of numerous cytokines and chemokines in genetically susceptible patients like those having HLA-B51 might lead to the development of Kikuchi-Fujimoto disease like in our case. The diagnosis of Kikuchi-Fujimoto disease is mostly based on the lymph node biopsy findings. However, FNAC is also a useful modality with an overall accuracy of 56 per cent.7 Histology shows apoptotic coagulation necrosis with karyorrhectic debris along with proliferation of histiocytes, plasmacytoid dendritic cells, and CD8+ T-cells in the absence of neutrophils. Immunohistochemistry predominantly shows CD8+ T-cells, histiocytes expressing myeloperoxidase, and lysozyme. Histiocytes in Kikuchi-Fujimoto disease also show positive staining with CD68.2
Common differentials of Kikuchi-Fujimoto disease are lymphoma, tuberculosis, and systemic lupus erythematosus. Lymphadenitis in systemic lupus erythematosus can be differentiated by the presence of haematoxylin bodies (amorphous aggregates of basophilic material) and DNA deposits in the blood vessel wall surrounding the necrotic foci.6 Kikuchi-Fujimoto disease, in contrast to lymphoma, will show relatively low mitotic rates, absent Reed-Sternberg cells, numerous histiocytes, preservation of nodal architecture, and polyclonal infiltrate. A diagnosis of tuberculosis is favoured when, in addition to necrotising lymphadenitis, there is proliferation of epithelioid histiocytes with granuloma formation and scattered giant cells.6 Kikuchi-Fujimoto disease usually undergoes spontaneous resolution within 4 to 6 months. There is no specific treatment for Kikuchi-Fujimoto disease, and supportive management is the mainstay of the treatment. It is commonly misdiagnosed, which often leads to a delay in diagnosis. Apart from its common association, Kikuchi-Fujimoto disease can be associated with Behcet disease, as seen in our case. As the disease is self-limiting and has a good prognosis, differentiating it from its relatively severe differentials is crucial for timely diagnosis and avoidance of exhaustive workup as well as aggressive treatment.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Kikuchi-Fujimoto disease: A review. Arch Pathol Lab Med. 2018;142:1341-6.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical outcome and predictive factors of recurrence among patients with Kikuchi’s disease. Int J Infect Dis. 2009;13:322-6.
- [CrossRef] [PubMed] [Google Scholar]
- Kikuchi-Fujimoto disease associated with neuro-Behçet’s disease. Clin Neurol Neurosurg. 2021;202:106541.
- [CrossRef] [PubMed] [Google Scholar]
- Kikuchi-Fujimoto disease: A comprehensive review. World J Clin Cases. 2023;11:3664-79.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Diagnosing Kikuchi disease on fine needle aspiration biopsy: a retrospective study of 44 cases diagnosed by cytology and 8 by histopathology. Acta Cytol. 2001;45:953-7.
- [CrossRef] [PubMed] [Google Scholar]




