
Translate this page into:
Langerhans cell histiocytosis in an adult female with atypical swellings
-
Received: ,
Accepted: ,
How to cite this article: Shukla P, Verma P, Kushwaha R, Verma S, Yadav G, Nazar AH. Langerhans cell histiocytosis in an adult female with atypical swellings. Indian J Dermatol Venereol Leprol 2021;87:254-9.
Sir,
Langerhans cell histiocytosis rarely affects adults and is a diagnostic challenge owing to its varied clinical presentations. Treatment and prognosis depend on the organs involved. Cutaneous involvement is one of the common presentations and sometimes the first symptom to appear, giving the dermatologists a pivotal role to play in early diagnosis and management.1 We hereby present the case of a young female with atypical swellings and crusted papules in seborrheic distribution.
A 25-year-old female presented to the dermatology out-patient department with a gradually progressive 8 × 8 cm soft swelling on nape of the neck, studded with papules and pustules, many covered with thin crust, for past 18–20 months [Figure 1]. Similar, but smaller, swellings were present on the axillae [Figures 2 and 3]. Papulopustules were also distributed on seborrheic areas of head and neck and a few lesions showed umbilication. She gave a history of similar swelling on the back 5 years ago that resolved spontaneously in 2 years. Loss of appetite and weight loss were present. The patient had received treatment for the same under the diagnosis of eczema, hidradenitis suppurativa, candidiasis and recurrent furuncles from multiple hospitals but was never relieved. There were no complaints of fever or any other systemic illnesses.
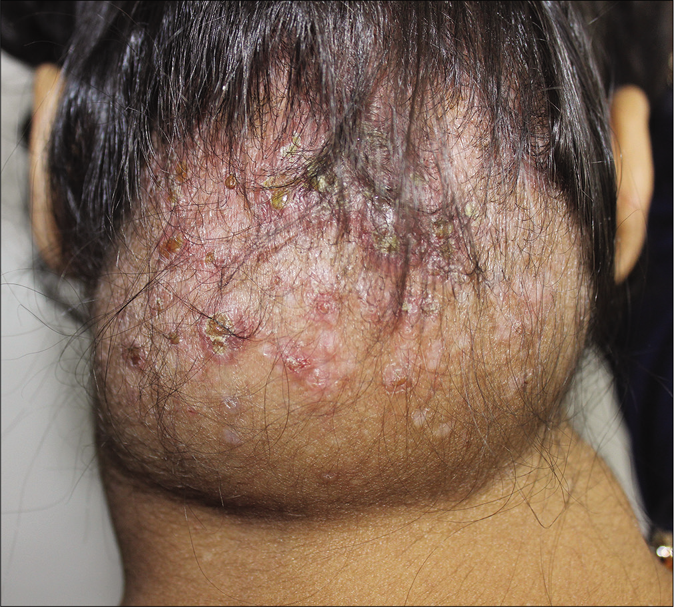
- Soft swelling on nape of neck studded with erythematous papules, many of them covered with yellowish crust
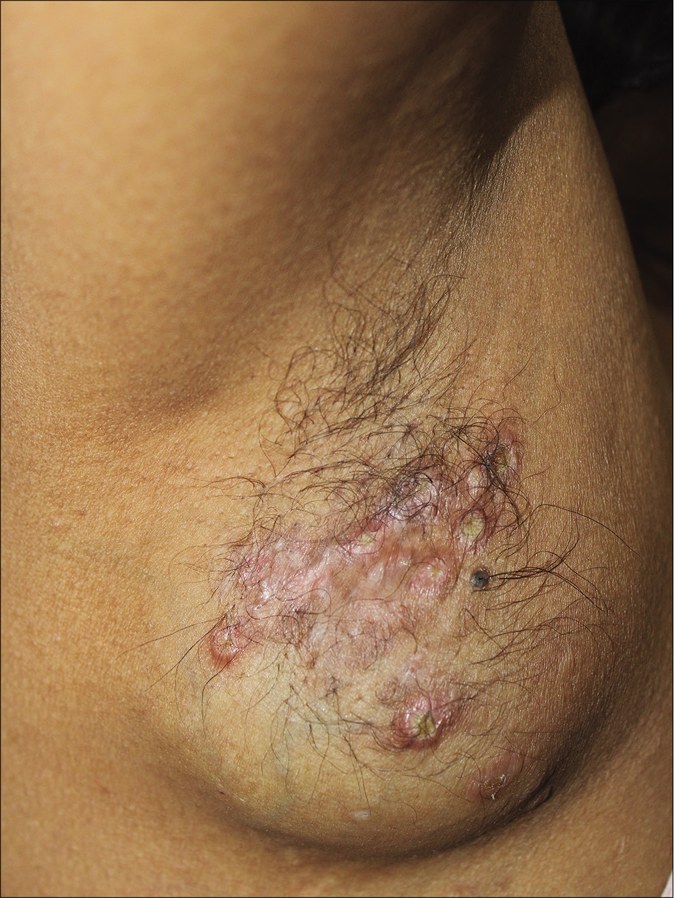
- Swelling on right axilla studded with erythematous papules covered with yellowish crust
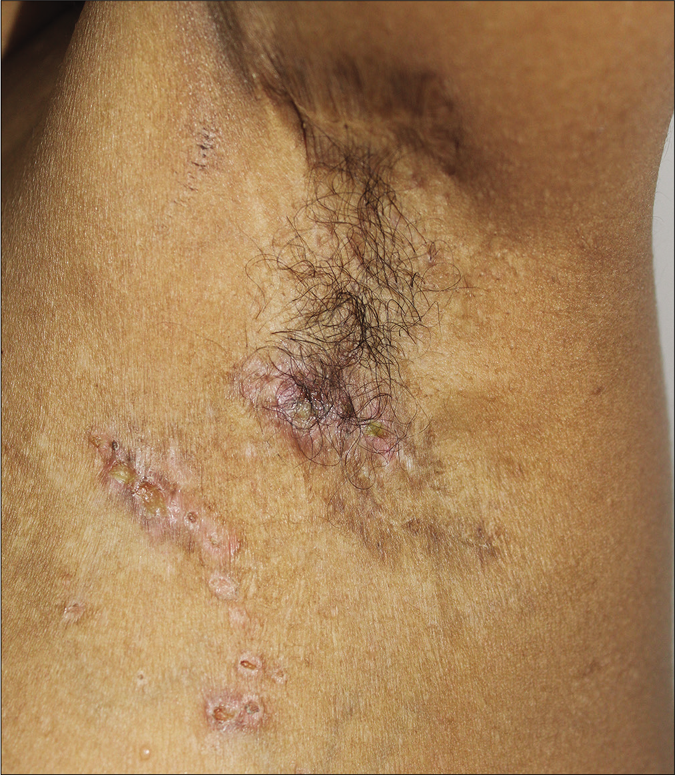
- Erythematous papules covered with yellowish crust in left axilla
Differential diagnoses considered were lymphoma, Langerhans cell histiocytosis, hidradenitis suppurativa, Rosai–Dorfmann disease, tuberculosis and histoplasmosis. The lesion on trunk was subjected to histopathology. Fine-needle aspiration cytology was done from swelling on the neck. Dense infiltrate in papillary dermis comprising of Langerhans cells having eosinophilic cytoplasm with coffee bean nuclei having vesicular chromatin and prominent nucleoli was found on histopathology which were positive for CD1a on immunohistochemistry [Figures 4-6]. Fine-needle aspiration cytology also showed similar picture of Langerhans cells [Figure 7].
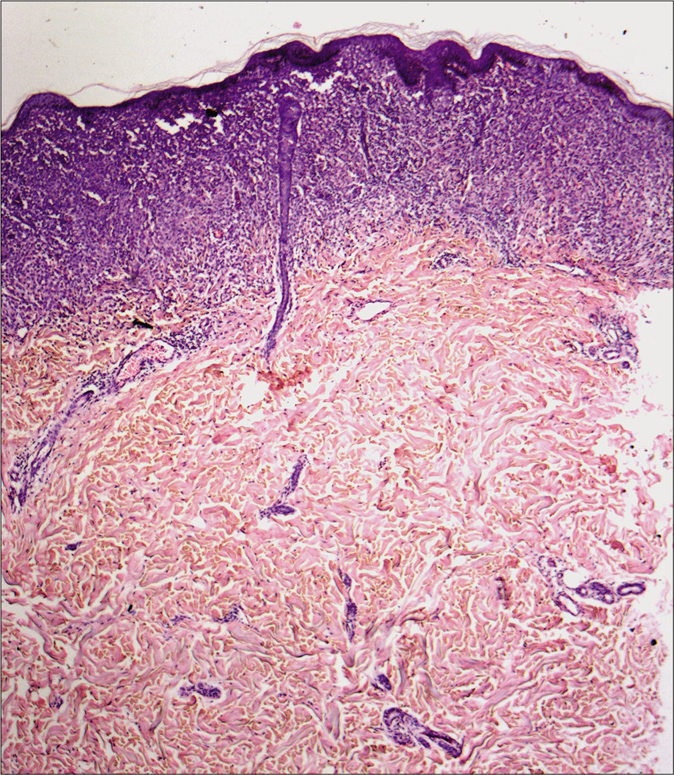
- Upper and mid dermal infiltrates (hematoxylin and eosin, ×40)
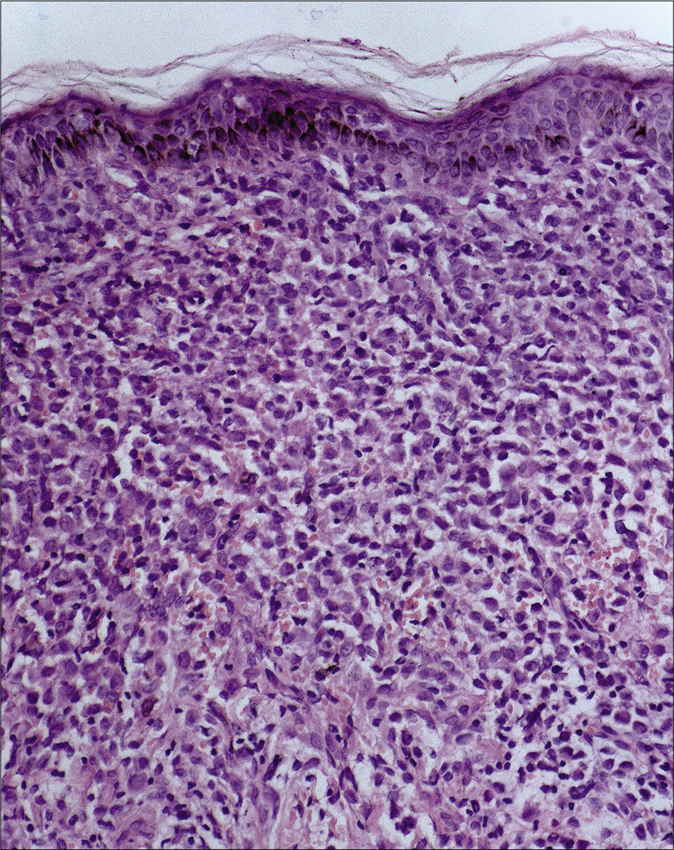
- Infiltration by abnormal Langerhans cells (round to polygonal cells with oval nuclei having longitudinal grooves resembling coffee bean) in dermis. Stroma also shows the presence of eosinophils (hematoxylin and eosin, ×200)
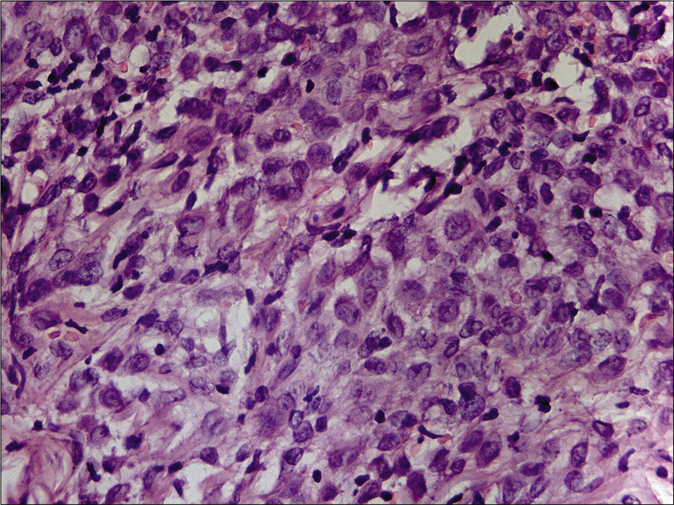
- Langerhans cells showing prominent nuclear grooves with fine chromatin and small nucleoli (hematoxylin and eosin, ×1000)
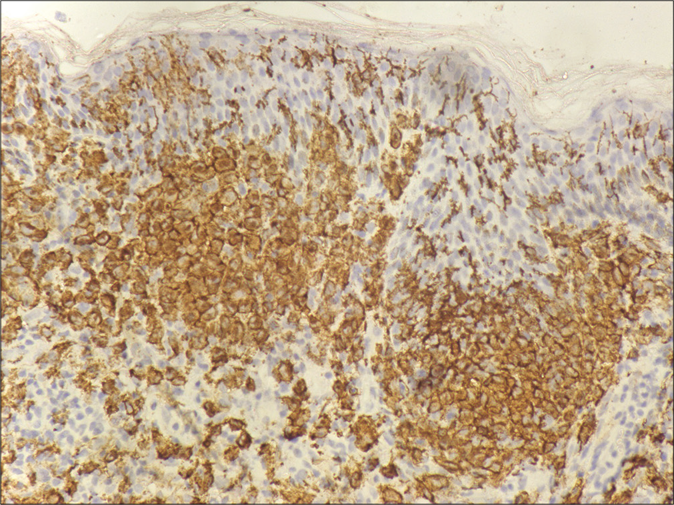
- Langerhans cells in dermis showing immunoreactivity to CD1a (×400)
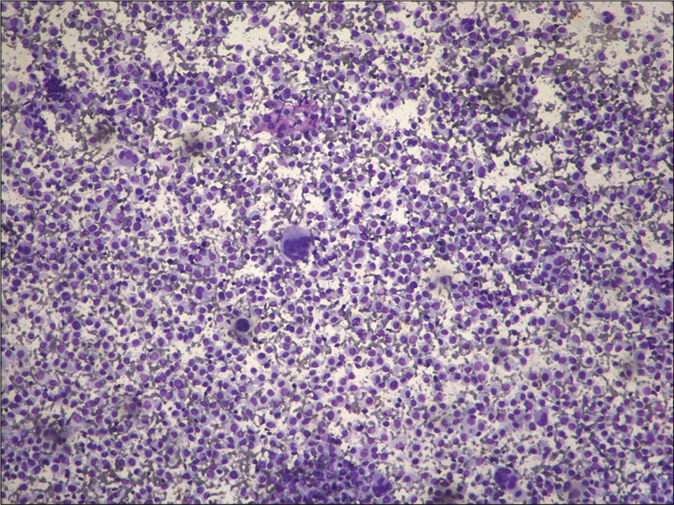
- Smear showing lymphocytes with a fair number of abnormal histiocytes (Leishman, ×100)
A provisional diagnosis of Langerhans cell histiocytosis was reached and patient was further investigated for assessment of other organs under the hemato-oncology division of the hospital. There were no abnormalities noted on X-ray skull and cervical spine. Ultrasound scan of abdomen also did not show any abnormality. But the positron emission tomography scan showed increased 18-fluorodeoxyglucose uptake identifying metabolically active disease at multiple sites, including soft tissues as well as bones, muscles, lymph nodes and lungs [Figures 8-11]. The hypermetabolic (maximum standardized uptake value 11.28) soft tissue thickening (~3.5 cmm) involved cutaneous and subcutaneous plane in nape of neck with cranio-caudal extension from occipital protuberance to level of C5 vertebra. Lesion extended into bilateral level V, bilateral level III cervical lymph nodal station and left carotid space without involvement of underlying occipital bone or cervical vertebrae. Diffuse area of honeycombing in bilateral lung fields predominantly in upper lobes with mild diffuse fluorodeoxyglucose avidity was noticed.
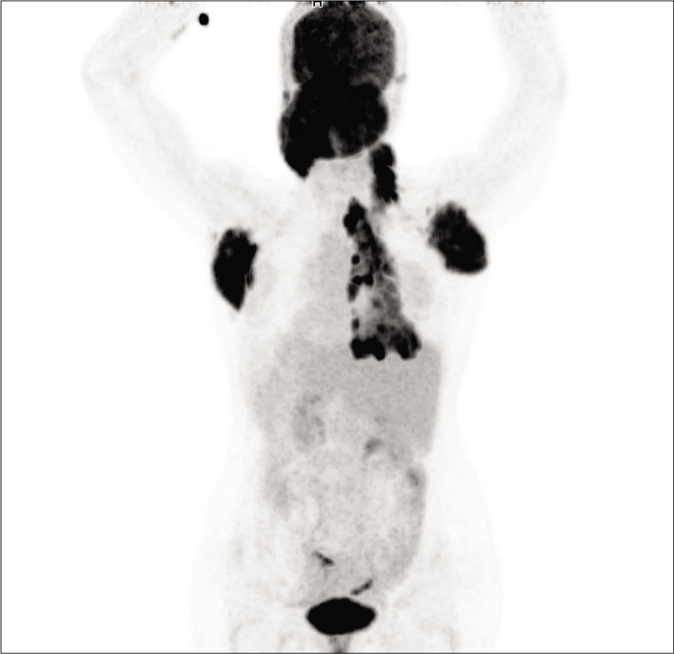
- Positron emission tomography (PET) scan showed increased 18-fluorodeoxyglucose (FDG) uptake in nape of neck, axilla and right posterior thoracic wall including skeletal lesion in rib and vertebra.
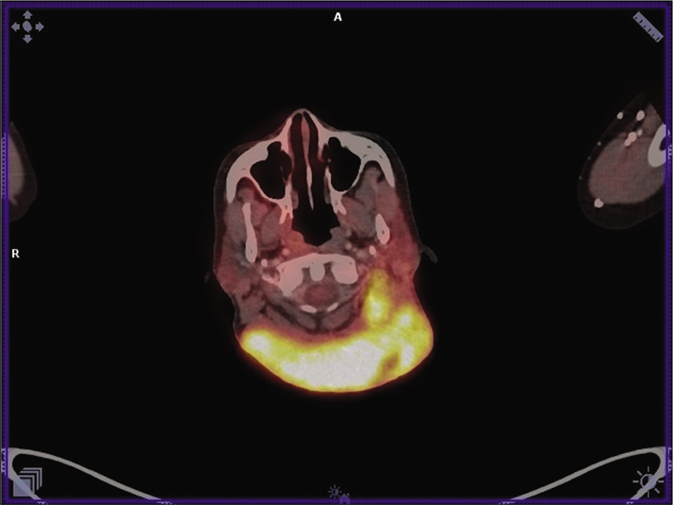
- PET scan showing increased 18-FDG uptake in lesions on the nape of neck and left cervical lymph nodes
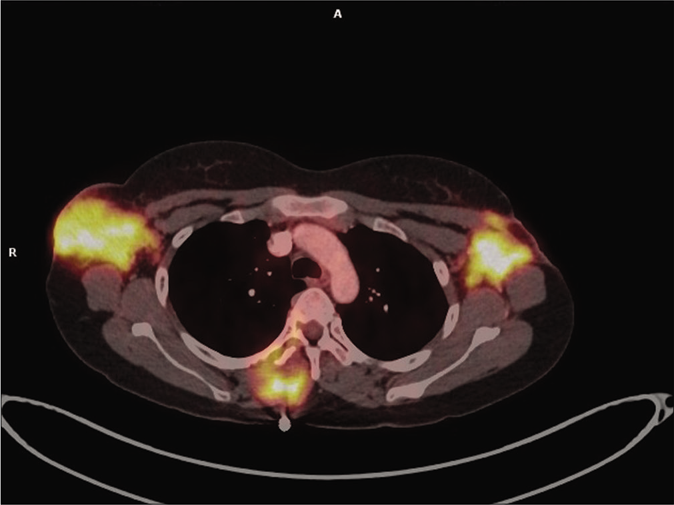
- PET scan showing increased 18-FDG uptake in bilateral axillary lymph nodes and lesion in the left axilla involving lateral border of pectoralis major and left latissimus dorsi muscles
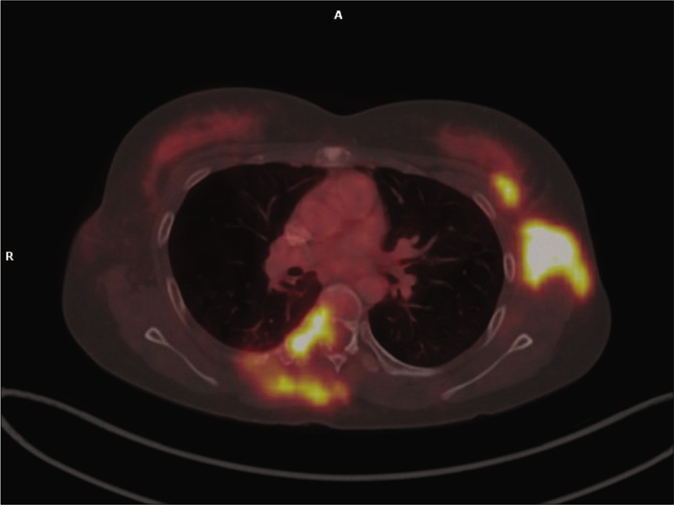
- PET scan showing increased 18-FDG uptake in D6 vertebra and right 6th rib
A diagnosis of multi-system Langerhans cell histiocytosis was made and the patient was treated with prednisolone (started at 40 mg/m2 and then tapered) and vinblastine (6 mg/m2, planned on receiving 12 cycles). A decrease in size of the large swelling was noted in her latest follow-up after 6 cycles of chemotherapy. Also, there were no new lesions on trunk and the axillary lesions started resolving with residual scarring [Figures 12 and 13].
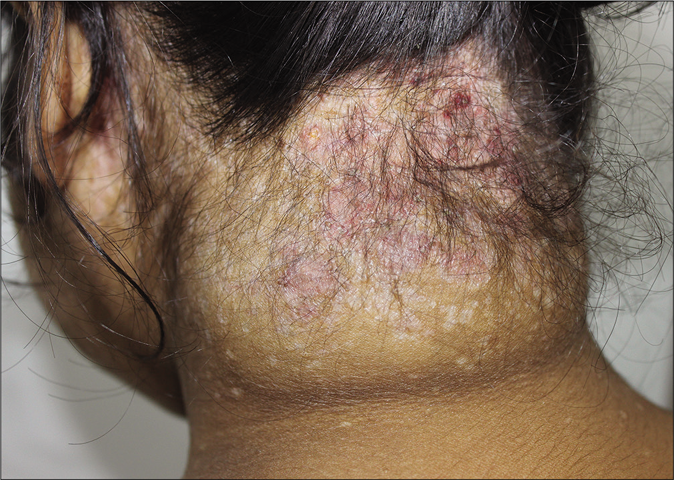
- Resolving neck lesions after chemotherapy
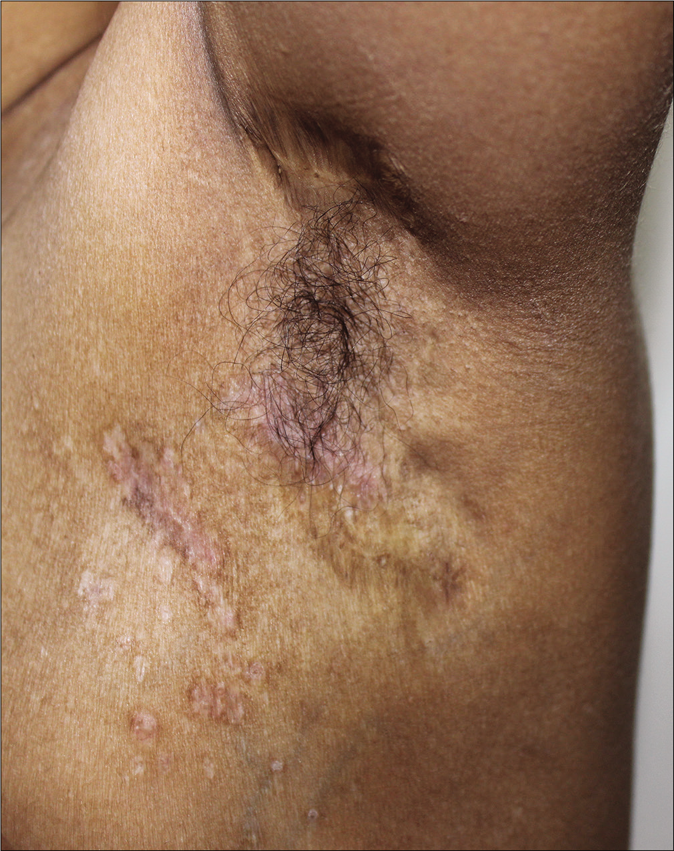
- Resolving left axillary lesions after chemotherapy
Langerhans cell histiocytosis (or histiocytosis X) is a rare clonal proliferative disorder of unknown etiology affecting mainly children. It is characterized by accumulation of Langerhans cells, with recent development suggesting that Langerhans cell histiocytosis arises from bone marrow-derived immature myeloid dendritic cells. The nature of the disease is debatable and has not been settled to reactive or neoplastic origin. It has recently been found to be associated with mutation in BRAF V600E hinting a neoplastic nature.2 The clinical presentation of Langerhans cell histiocytosis is highly variable, ranging from single bony or cutaneous lesion to multisystem involvement. It is classified on the basis of extent of disease into single system and multisystem disease.3 Multisystem disease is further divided into low-risk group (without involvement of organ) and high-risk group (involvement of liver, spleen and bone marrow) and different management strategies are required for different groups, owing to bad prognosis in high-risk group patients.
To find an adult affected with Langerhans cell histiocytosis is even rare, with an annual incidence being approximately 1–2 per million.4 Bone, followed by skin, is the most commonly involved organ and cutaneous manifestations are the first to appear in up to 80% patients.1 The lesions may present as papules, pustules, vesicles, petechiae, purpura or nodules, involving mainly scalp, face, trunk and intertriginous areas. Many unusual presentations have been reported like diffuse molluscum contagiosum-like umbilicated papules, ulcerative plaque and those mimicking lichen nitidus.5-7 Our patient had pleomorphic lesions later proved to be of histiocytic origin, with the typical papules and crusts in seborrheic distribution and the atypical large swelling at the nape of the neck. The uncommon presentation and the rarity of the condition in adults caused it to evade detection for months.
Recently, 18-fluorodeoxyglucose positron emission tomography/computerized tomography (FDG-PET/CT) has emerged as a novel tool for the evaluation of disease extent in multi-system Langerhans cell histiocytosis. It offers single diagnostic technique to screen the whole body simultaneously and identify organs involved in disease process. Albano et al. in their study evaluated 17 patients of histologically proven case of Langerhans cell histiocytosis and detected 36 positive lesions out of total 37 lesions.8 Authors concluded that this modality is more useful for evaluating the condition when compared to conventional imaging, except in pulmonary cases and it can be used both for staging and restaging purposes, as was also evident in our case. Interestingly, our patient had bony involvement in sites not otherwise clinically symptomatic (right 5th, 6th, 7th ribs posteriorly and adjacent D5-D7 vertebrae), whereas the occipital bone and cervical vertebrae underlying the painless soft tissue swelling that mimicked bone lesion were found to be uninvolved. These and a few other metabolically active lesions could only be identified upon positron emission tomography scan, thus emphasizing its role. Furthermore, despite being asymptomatic clinically, she also had involvement of lungs and vertebrae, which would have been missed if not for the above, thereby causing under-staging and under-treatment.
Histopathology shows Langerhans cells that are 12 to 15 μm in diameter with abundant eosinophilic cytoplasm. The nuclei of these cells are irregular with prominent folds and grooves (coffee bean appearance), fine chromatin and indistinct nucleoli. The immunophenotype characteristically shows expression of CD1a, S100 protein and langerin (CD207). On electron microscopy, “tennis-racket” shaped cytoplasmic structures, known as Birbeck granules, are observed. Diagnosis can also be made on fine-needle aspiration cytology sample if the patient has swellings as in this case. This may help in rapid diagnosis.9
Treatment of Langerhans cell histiocytosis depends upon the extent and site of involvement and thus, the staging. Systemic therapy is required for multifocal single-system and multisystem Langerhans cell histiocytosis (with or without organ involvement). Vinblastine/prednisolone has been used widely in pediatric patients with satisfactory results but owing to its rare occurrence in adults, its efficacy is not established.6
Cytarabine, cladribine and etoposide have been tried in addition to conventional modalities like methrotrexate, azathioprine and thalidomide.6 Also, radiotherapy has a role in selective cases of adults, as opposed to pediatric cases.6 More recently, vemurafinib, a tyrosine-kinase inhibitor, has been used albeit off-label, in a BRAF V600E positive, multi-refractory disease.10
This patient received systemic therapy in view of multisystem disease. Long-term follow-up is required to lookout for relapse which is a dreaded risk in the adult population.11 The overall prognosis depends upon the extent of disease, with localized disease having the best prognosis and a 5-year survival rate of 100%.12 A high index of suspicion is required especially in the adults so as not to miss the diagnosis of this rare condition, more so when the presentation is atypical.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Adult onset of multisystem Langerhans cell histiocytosis with skin and lymph node involvement. Postepy Dermatol Alergol. 2015;32:225-8.
- [CrossRef] [PubMed] [Google Scholar]
- Recent advances in Langerhans cell histiocytosis. Pediatr Int. 2014;56:451-61.
- [CrossRef] [PubMed] [Google Scholar]
- Management of adult patients with Langerhans cell histiocytosis: Recommendations from an expert panel on behalf of Euro Histio Net. Orphanet J Rare Dis. 2013;8:72.
- [CrossRef] [PubMed] [Google Scholar]
- 55 year old man with ulcers in inguinal fold and intergluteal cleft found to have systemic Langerhans cell histiocytosis. JAAD Case Rep. 2018;4:837-40.
- [CrossRef] [PubMed] [Google Scholar]
- Langerhans cell histiocytosis: A case report with unusual cutaneous manifestation. Adv Biomed Res. 2018;7:102.
- [CrossRef] [PubMed] [Google Scholar]
- Molluscum contagiosum like presentation of langerhans cell histiocytosis: A case and review. Pediatr Dermatol. 2017;34:e288-9.
- [CrossRef] [PubMed] [Google Scholar]
- Langerhans cell histiocytosis mimicking lichen nitidus with bone involvement. Australas J Dermatol. 2017;58:231-3.
- [CrossRef] [PubMed] [Google Scholar]
- Role of 18F FDG PET/ CT in patients affected by Langerhans cell histiocytosis. Jpn J Radiol. 2017;35:574-83.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosis of Langerhans cell histiocytosis on fine needle aspiration cytology: A case report and review of the cytology literature. Patholog Res Int 2011:1-5.
- [CrossRef] [PubMed] [Google Scholar]
- Access to molecular guided therapy for Langerhans cell histiocytosis patients. J Am Acad Dermatol. 2015;73:e31.
- [CrossRef] [PubMed] [Google Scholar]
- Langerhans cell histiocytosis: Diagnosis, natural history, management, and outcome. Cancer. 1999;85:2278-90.
- [CrossRef] [Google Scholar]
- Langerhans cell histiocytosis in adults, Report from the International Registry of the Histiocyte Society. Eur J Cancer. 2003;39:2341-8.
- [CrossRef] [Google Scholar]




