Translate this page into:
Langerhans cell histiocytosis of skin: A clinicopathologic analysis of five cases
2 Departments of Dermatology, Govt. Medical College, Chandigarh, India
Correspondence Address:
Harsh Mohan
Department of Pathology, Govt. Medical College, Sector 32 A, Chandigarh
India
| How to cite this article: Punia RS, Bagai M, Mohan H, Thami G P. Langerhans cell histiocytosis of skin: A clinicopathologic analysis of five cases. Indian J Dermatol Venereol Leprol 2006;72:211-214 |
Abstract
Background and Aims: Langerhans cell histiocytosis (LCH) is a rare proliferative disorder of histiocytes characterized by a proliferation of abnormal and clonal Langerhans cells. We retrospectively studied clinicopathologic features of this disorder in five cases. Methods: Clinical and histopathological findings of five cases of cutaneous LCH were reviewed based on the hospital records. Results: The age of patients ranged from 28 days to 5 years and M: F ratio was 1:1.5. Clinically, the diagnoses suggested were histiocytosis, varicella, transient neonatal pustular melanosis, keloid, sarcoidosis, seborrheic keratosis and LCH. The most common type of skin lesion was a generalized papular lesion. Histologically, all cases showed aggregates of large mononuclear histiocytes (Langerhans cells) with reniform, irregular, cleaved nuclei and abundant eosinophilic cytoplasm. There was multi-systemic involvement in two patients and single-system involvement in three patients. Conclusion: Cutaneous lesions may be the sole presenting feature of LCH. Diagnosis is based on demonstration of S-100 positive histiocytes.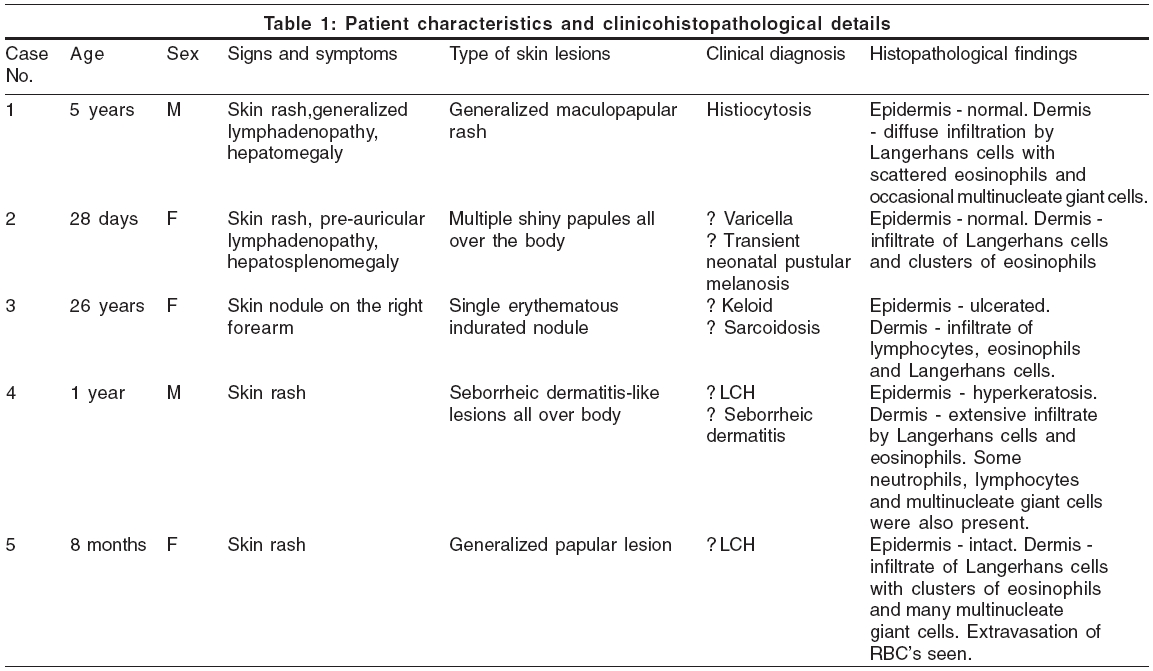
 |
|
 |
|
Introduction
Langerhans cell histiocytosis (LCH) is a proliferative disorder of histiocytes that is characterized by heterogenous clinical manifestations and an unpredictable course.[1] LCH includes diseases previously designated as histiocytosis X, eosinophilic granuloma, Letterer-Siwe disease, Hand-Schuller-Christian disease, Hashimoto-Pritzker syndrome, self-healing reticulocytosis, pure cutaneous histiocytosis, Langerhans cell granulomatosis, type II histiocytosis and nonlipid reticuloendotheliosis.[2] It is characterized by a proliferation of abnormal and clonal Langerhans cells in one or more body organs such as the skin, bone, lymph node, lungs, liver, spleen and bone marrow.[3] The disease can present at any age, though commonly it presents in infancy or early childhood, often with cutaneous lesions. Prognostically, it is a confounding disorder with a wide spectrum of outcomes ranging from spontaneous remissions to metastasis and death.[4]
We present a retrospective clinicopathologic analysis of five patients with cutaneous LCH diagnosed in our laboratory over the past 5 years.
Methods
The present study included five cases of LCH diagnosed in our laboratory between January 1999 and December 2004. The diagnosis was established on biopsy. All the biopsies received were routinely processed and stained with routine H and E stain and S-100 immunostain. The clinical data was retrieved from the surgical biopsy forms and the case files of the patient. The following clinical parameters were evaluated: age at diagnosis, signs and symptoms (presence and type of skin lesions, lymphadenopathy, bone pain and enlargement of the liver and spleen), site of biopsy, single/multi-system disease, other investigations (e.g., hemogram, radiographs of skull and chest, liver function tests) and follow-up of the patient (wherever available).
Results
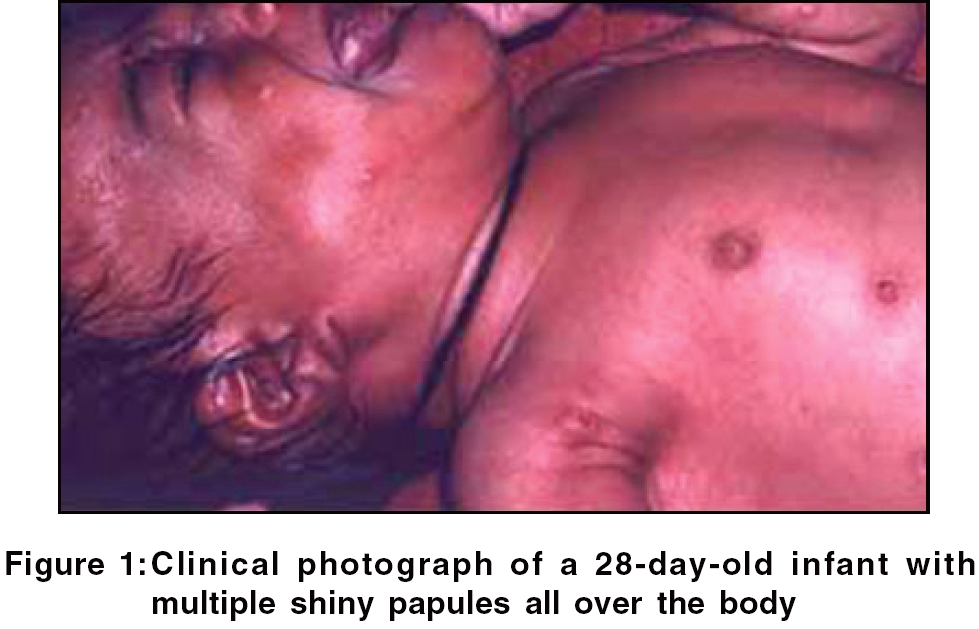
The age of patients ranged from 28 days to 26 years; three out of the five patients presented in infancy. The male to female ratio was 1:1.5. In all patients, the skin was involved and skin lesions were the presenting feature; the most common type was a generalized papular lesion (three patients) [Figure - 1], followed by a seborrheic dermatitis type of lesion and a single erythematous nodule (one patient each). There was multi-systemic involvement in two patients and single-system involvement in three. Patients with multisystemic involvement did not show any systemic symptoms but had lymphadenopathy and enlargement of the liver and spleen. Hemogram, liver function tests and radiographs of the chest and skull were normal in all the cases. The salient features are summarized in [Table - 1].
On histopathology, the epidermis was intact in four cases, but in one case (case no. 3), which presented as a solitary nodule, the overlying epidermis was ulcerated. The dermis showed aggregates of Langerhans cells, which are large mononuclear histiocytes with reniform, irregular, cleaved nuclei and abundant eosinophilic cytoplasm [Figure - 2]. These Langerhans cells were admixed with other inflammatory cells, particularly eosinophils. The eosinophils were seen diffusely scattered along with other inflammatory cells in three patients (cases nos. 1, 3 and 4), while they were seen in clusters in two others (cases nos. 2 and 5). In addition, three patients (cases nos 1, 4 and 5) showed the presence of multinucleate giant cells. The Langerhans cells were positive for S-100.
Discussion
Lichtenstein, in 1953, coined the term histiocytosis-X to describe a group of syndromes (Hand-Schuller-Christian disease, Letterer-Siwe disease and eosinophilic granuloma) with seemingly unrelated clinical features but whose pathological findings were characterized by the infiltration of involved tissues with large number of unusual histiocytes.[5] Subsequently, these histiocytes were found similar to Langerhans cells normally present in the skin and other epithelia; thereafter, the term Langerhans cell histiocytosis (LCH) was used.[6]
The disease has an incidence of 1 per 1,000,000 in children younger than 15 years, with a peak incidence between the age of 1 and 4 years. However, it should not be considered a disease that is confined to children.[7],[8] LCH is a clonal neoplastic disorder. The etiopathogenesis is still not known,[9] but the role of Epstein-Barr virus (EBV) is being studied, as EBV expression was found in LCH using in situ hybridization and immunofluorescence.[10] The disease may involve one or more body organ systems or tissues, such as bone, skin/mucous membranes, lung, lymph nodes, hypothalamus/posterior pituitary gland, liver and various soft tissues including the testes.[9] The most common presenting features include fever, hepatosplenomegaly, skin lesions, lymphadenopathy and bone marrow involvement.[8]
In an extensive study of 314 patients of histologically proven LCH at Mayo Clinic,[9] multisystem LCH was found in 30% of cases; isolated bone lesions in 36%, with the skull and proximal femur being the most common sites. Other non-osseous single-system LCH (34%) was found in the pulmonary, mucocutaneous, thalamic axis and lymphatic systems. Cutaneous involvement in LCH was seen in 50% of the cases and may be the presenting feature. However, isolated cutaneous lesions are relatively uncommon in adults as compared to children.[11] The most common initial skin lesions include vesiculopustules, seborrheic dermatitis-like rash, mucosal lesions (erosions, petechiae and granulomas), erythematous papules, nodular ulcerative lesions and generalized petechiae.[4] The most common mucous membranes involved are the oral and genital mucosa. Of our five cases, two had multi-system disease, while in three cases the disease was confined to the skin.
The diagnosis of LCH is based on the presence of Langerhans cells, along with histiocytes and eosinophils, on histopathological examination. Three types of histological reactions have been described in LCH: proliferative, granulomatous and xanthomatous. The proliferative reaction is characterized by the presence of an extensive infiltrate of histiocytes along with an admixture of lymphocytes and scattered eosinophils. The infiltrate usually lies close to the epidermis and may result in ulceration and crusting. The granulomatous reaction shows extensive aggregates of histiocytes and clusters of eosinophils extending deep into the dermis. Occasional multinucleate giant cells are also seen. The xanthomatous reaction is relatively uncommon and shows numerous foam cells along with histiocytes and eosinophils.[12] In our series, cases 1, 3 and 4 showed the proliferative kind of lesions, while cases 2 and 5 showed granulomatous histiocytosis.
Cutaneous lesions may be the sole presenting feature in LCH; the diagnosis is based on the presence of characteristic S-100 positive Langerhans cells.
| 1. |
Kusumkumary P, Kumari PT, Chellam VG, James FV, Nair KM. Langerhans cell histiocytosis in children less than 2 years of age. Indian Pediatr 1999;36:29-36.
[Google Scholar]
|
| 2. |
Chu T, D'Angio GJ, Favara B, Ladisch S, Nesbit M, Pritchard J. The writing group of the histiocytic society: Histiocytic syndromes in children. Lancet 1987;1:208-9.
1987;1:208-9.'>[Google Scholar]
|
| 3. |
Bernstrand C, Carstensen H, Jakobsen B, Svejgaard A, Henter J, Olerup O. Immunogenetic heterogeneity in single-system and multisystem Langerhans cell histiocytosis. Pediatr Res 2003;54:30-6.
[Google Scholar]
|
| 4. |
Stein SL, Paller AS, Haut PR, Mancin AJ. Langerhans cell histiocytosis presenting in the neonatal period: A retrospective case series. Arch Pediatr Adolesc Med 2001;155:778-83.
[Google Scholar]
|
| 5. |
Lichtenstein L, Histiocytosis X. Integeration of eosinophilic granuloma of bone, Letterer-Siwe disease and Hand-Schuler-Christian disease, as related manifestations of a single nosologic entity. Arch Pathol 1953;56:84-102.
[Google Scholar]
|
| 6. |
Nezelof C, Basset F, Rousseau MF. Histiocytosis X: Histogenic arguments for a Langerhans cell origin. Biomedicine 1973;18:365-71.
[Google Scholar]
|
| 7. |
Favara BE, Mac Carthy RC, Mierau GW. Histiocytosis X. Human Pathol 1983;14:663-76.
[Google Scholar]
|
| 8. |
Goyal A, Rani S, Singh T, Choudhary P, Dubey AP. Childhood histiocytosis: A review of twenty two cases. Indian Pediatr 1998;35:151-6.
[Google Scholar]
|
| 9. |
Howarth DM, Gilchrist GS, Mullan BP, Wiseman GA, Edmonson JH, Schomberg PJ. Langerhans cell histiocytosis: Diagnosis, natural history, management and outcome. Cancer 1999;85:2278-90.
[Google Scholar]
|
| 10. |
Shimakage M, Sasagawa T, Kimura M, Shimakage T, Seto S, Kodama K, et al . Expression of Epstein-Barr virus in Langerhans cell histiocytosis. Hum Pathol 2004;35:862-8.
[Google Scholar]
|
| 11. |
Singh A, Prieto VG, Czelusta A, McClain KL, Duvic M. Adult Langerhans cell histiocytosis limited to the skin. Dermatology 2003;207:157-61.
[Google Scholar]
|
| 12. |
Burgdorf WHC. The Histiocytosis. In: Elder D, Elenitsas R, Jaworsky C, Johnson Jr. B, editors. Lever's Histopathology of the Skin. 8th ed. Lippincott-Raven: Philadelphia; 1997. p. 591-616.
[Google Scholar]
|
Fulltext Views
10,382
PDF downloads
3,259





![[Figure - 1]](#fig_ijdvl_2006_72_3_211_25782_2.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2006_72_3_211_25782_1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2006_72_3_211_25782_3.jpg){kind=link}