
Translate this page into:
Late-onset hereditary sensory and autonomic neuropathy type 2B caused by novel compound heterozygous mutations in FAM134B presenting as chronic recurrent ulcers on the soles
Corresponding author: Dr. Zhi-Miao Lin, Department of Dermatology, Peking University First Hospital, Beijing Key Laboratory of Molecular Diagnosis on Dermatoses and National Clinical Research Center for Skin and Immune Diseases, Beijing 100034, China. zhimiaolin@bjmu.edu.cn
-
Received: ,
Accepted: ,
How to cite this article: Luo Z, Wang H, Zhao Y, Liu J, Chen Y, Lin Z, et al. Late-onset hereditary sensory and autonomic neuropathy type 2B caused by novel compound heterozygous mutations in FAM134B presenting as chronic recurrent ulcers on the soles. Indian J Dermatol Venereol Leprol 2021;87:455.
Sir,
A 38-year-old man from a nonconsanguineous Chinese family presented with a 23-year history of recurrently flared erythema, swelling and pain on the toes and forefeet. Twenty years ago, mutilating ulceration with discharges of sequestrum subsequent to the flared lesion were present on his soles; later the ulceration became painless and healed spontaneously with scars. He had difficulty in walking three years before his presentation and was diagnosed as leprosy and peripheral neuropathy that showed poor response to the treatments. Examination showed deformity of multiple toes and plantar ulceration on the right forefoot [Figure 1a], associated with onychodystrophy, localized plantar hyperkeratosis and scars on both feet. All muscle groups and tendon reflexes were normal. Roentgen-ray revealed pedal acro-osteolysis and resorption of the terminal phalanges and metatarsal bones of the first and second toes on both feet [Figure 1b]. Electromyography revealed mildly slow sensory and motor nerve conduction velocities as well as decreased sensory nerve action potential in the lower limbs [Table 1]; however, no myogenic involvement was present. Nerve biopsy was denied. Laboratory tests including complete blood cell count, biochemical profiles, serum creatine kinase-MB, immunoglobulin, complements and autoimmune antibodies were within the normal range or negative. During a four-year follow-up, progressive osteolysis on the left middle finger was present. The patient showed poor response to palliative treatments.
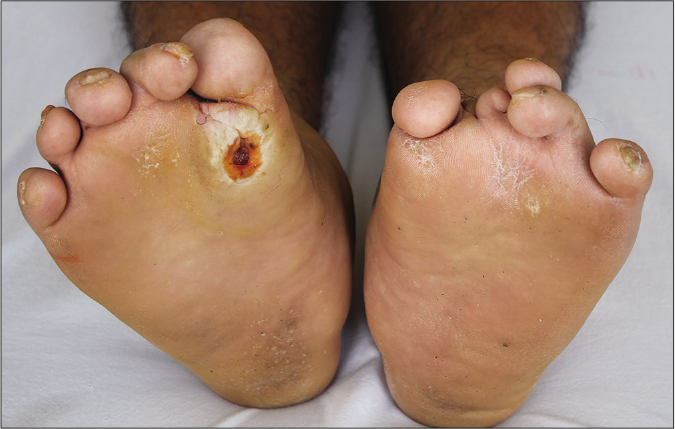
- Malformation of both feet associated with localized plantar hyperkeratosis and scars, and ulceration on the right forefoot
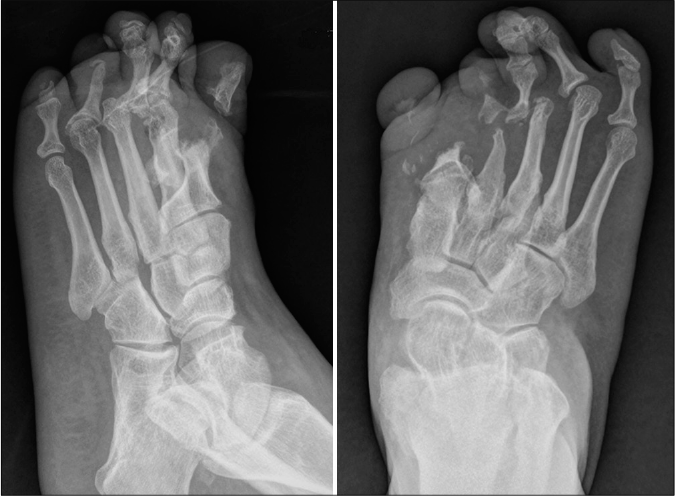
- Radiology reveals acro-osteolysis and resorption of the terminal phalanges and metatarsal bones of the first and second toes
| Site | Distal latency (ms) | Amplitude | Velocities(m/S) |
|---|---|---|---|
| Sensory nerve conduction study Median, R | |||
| Thumb | 1.96 | 13.7 µV | 45.9 |
| Middle finger | 2.34 | 34.2 µV | 59.8 |
| Ulnar, R | |||
| Wrist | 2.42 | 27.0 µV | 49.6 |
| Sural, R | 2.82 | 7.8 µV | 46.1 |
| Sural, L | 3.32 | 7.6 µV | 36.1 |
| Superficial peroneal nerves | |||
| Superficial peroneal nerve, L | 1.92 | 9.6 µV | 52.1 |
| Superficial peroneal nerve, R | 3.30 | 8.1 µV | 42.4 |
| Motor nerve conduction study | |||
| Median, L | 3.50 | 11.34 mV | 57.1 |
| Ulnar, L | 2.86 | 13.57 mV | 83.9 |
| Peroneal, L | 5.95 | 2.0 mV | 50.4 |
| Peroneal, R | 4.5 | 2.24 mV | 42.2 |
The proband’s 30-year-old brother and 47-year-old sister had similar conditions starting in their second decade; however, both patients were absent for finger-osteolysis till the present submission. All three affected siblings had progressive pedal acro-osteolysis but lacked distal numbness and weakness in the extremities, and their temperature and touch sensation were slightly impaired. The sister had the most severe acro-osteolysis. All their siblings including another three normal women had normal birth and normal developmental milestones. They had normal sensations, proprioception and vibration, and pupillary reaction to light, and were absent for autonomic symptoms including cough, gastro-oesophageal reflux, postural hypotension, urinary incontinence, excessive sweating, hypesthesia, as well as hypalgesia.
Informed consent was obtained from each participated individual of the family. Whole-exome sequencing using the genomic DNA extracted from peripheral blood of the proband revealed two novel heterozygous mutations in FAM134B (c.1009C>T, p.Arg337*; c.259_266del, p.Leu87Glufs*51) which were confirmed by Sanger sequencing [Figures 2a and b]. The other two affected siblings also harbored the same mutations. Their father as well as the proband’s son were carriers of c.1009C>T and their mother was a carrier of c.259_266del [Figures 2a and b], respectively.
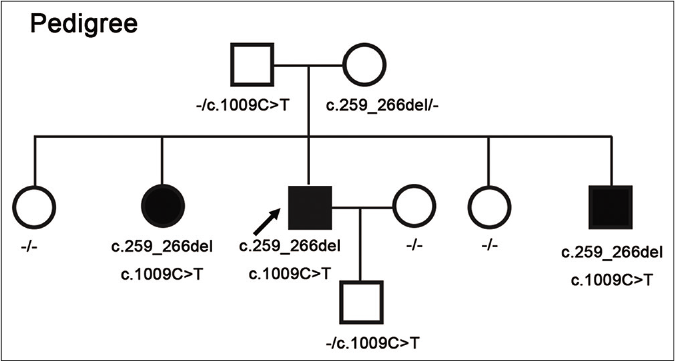
- Pedigrees of the described family with hereditary sensory and autonomic neuropathy type 2B. Filled boxes indicate affected individuals with compound heterozygous mutations in FAM134B (c.1009C>T, p.Arg337*; c.259_266delCTGAGCTG, p.Leu87Glufs*51). Their mother was a carrier of c.259_266del and their father as well as the proband’s son were carriers of c.1009C>T, respectively
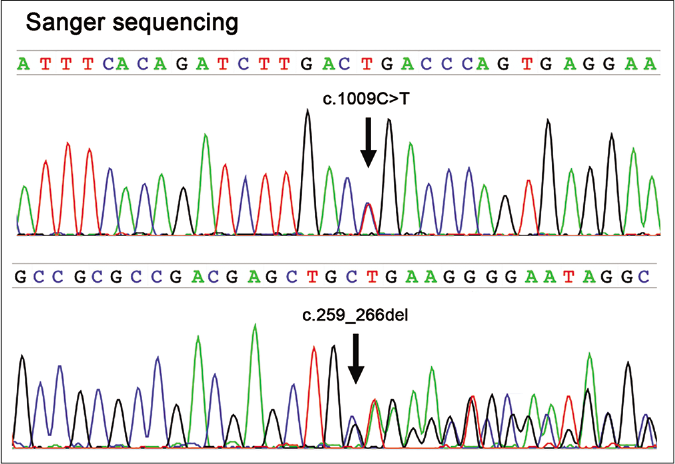
- Sanger sequencing of the FAM134B gene (NM 001034850) reveals compound heterozygous mutations (c.1009C>T, p.Arg337*; c.259_266delCTGAGCTG, p.Leu87Glufs*51) in the proband
A diagnosis of hereditary sensory and autonomic neuropathy type 2B was confirmed for the family. Compared with other subtypes of hereditary sensory and autonomic neuropathies, patients with hereditary sensory and autonomic neuropathy type 2, with highly variable progression, always have progressively reduced sensation to pain, temperature and touch in limbs with normal mental development and minimal autonomic dysfunction, their muscle weakness and wasting are usually absent or only mild. Hereditary sensory and autonomic neuropathy type 2 is comprised of four subtypes (2A, 2B, 2C and 2D). Hereditary sensory and autonomic neuropathy type 2B is a rare autosomal recessive disorder characterized by early-onset, pronounced sensory and autonomic dysfunction with or without distal motor abnormality.1 Patients with hereditary sensory and autonomic neuropathy type 2B usually present with chronic acral-osteomyelitis, recurrent infection, leading to painless mutilating ulceration and distal deformity1,2 which, sometimes, may be the main symptoms for the patients presenting to the dermatologist. Other cutaneous associations include excessive sweating, recurrent flares of erythema, nail dystrophy or loss, palmoplantar hyperkeratosis, widespread scarring, hypopigmentation and painful callus and fixed joint deformities.1,2 However, those symptoms are atypical and nonspecific. When a patient presents with recurrent chronic acro-osteomyelitis and one or more cutaneous features mentioned above, the diagnosis of hereditary sensory and autonomic neuropathy type 2B should be considered and a comprehensive evaluation of neurological manifestations or genetic testing should be performed.
Hereditary sensory and autonomic neuropathy type 2B is caused by recessive mutations of FAM134B, that encodes for a cis-Golgi transmembrane protein, also known as JK1, and renamed as reticulophagy regulator 1 by Human Genome Organization Gene Nomenclature Committee (http://www. genenames. org/) recently.3 Mutations in FAM134B lead to alterations of the cis-Golgi compartment that is crucial for the distribution and processing of proteins and lipids. As a result, loss of function in FAM134B results in apoptosis and degeneration of sensory and autonomic neurons. Till now, nine mutations in FAM134B (c.433C>T, c.471C>T, c.646G>A, c.873 + 2T>C, c.926C>G, c.17del, c.826del, c.1009C>T, c.259_266del) have been reported underlying hereditary sensory and autonomic neuropathy type 2B; all these mutations are predicted to be loss-of-function.1-5 These patients are novel entities with a relatively late onset of the symptoms associated with atypical cutaneous alterations, and with novel compound heterozygous truncated mutations in FAM134B. Owing to the rarity of hereditary sensory and autonomic neuropathy type 2B, it is necessary to have further study and accumulate more cases to unravel the genotypephenotype correlation of FAM134B-associated hereditary sensory and autonomic neuropathy type 2B and the cutaneous complications in the future.
Treatment strategies are still challenging. Preventive and symptomatic management including padded shoe-wear as well as gait and posture modification may be helpful.
The main limitations of the present work are that we have not studied the mechanisms of present compound heterozygous mutations.
Acknowledgment
The authors sincerely thank the patients and their family participating the present work.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Mutation in FAM134B causing hereditary sensory neuropathy with spasticity in a Turkish family. Muscle Nerve. 2014;49:774-5.
- [CrossRef] [PubMed] [Google Scholar]
- Mutation in FAM134B causing severe hereditary sensory neuropathy. J Neurol Neurosurg Psychiatry. 2012;83:119-20.
- [CrossRef] [PubMed] [Google Scholar]
- ETREG1 (FAM134B): A new player in human diseases: 15 years after the discovery in cancer. J Cell Physiol. 2018;233:4479-89.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in FAM134B, encoding a newly identified Golgi protein, cause severe sensory and autonomic neuropathy. Nat Genet. 2009;41:1179-81.
- [CrossRef] [PubMed] [Google Scholar]
- Frequency of mutations in the genes associated with hereditary sensory and autonomic neuropathy in a UK cohort. J Neurol. 2012;259:1673-85.
- [CrossRef] [PubMed] [Google Scholar]




