Translate this page into:
Livedoid vasculopathy: A review of pathogenesis and principles of management
2 Department of Dermatology, Command Hospital, Kolkata, West Bengal, India
3 Department of Dermatology, Command Hospital, Pune, Maharashtra, India
Correspondence Address:
Biju Vasudevan
Department of Dermatology, INHS Asvini, Near RC Church, Colaba, Mumbai - 400 005, Maharashtra
India
| How to cite this article: Vasudevan B, Neema S, Verma R. Livedoid vasculopathy: A review of pathogenesis and principles of management. Indian J Dermatol Venereol Leprol 2016;82:478-488 |
Abstract
Livedoid vasculopathy is a rare cutaneous disease manifesting as recurrent ulcers on the lower extremities. The ulceration results in atrophic, porcelain white scars termed as atrophie blanche. The pathogenesis is yet to be understood with the main mechanism being hypercoagulability and inflammation playing a secondary role. The important procoagulant factors include protein C and S deficiency, factor V Leiden mutation, antithrombin III deficiency, prothrombin gene mutation and hyperhomocysteinemia. Histopathology of livedoid vasculopathy is characterized by intraluminal thrombosis, proliferation of the endothelium and segmental hyalinization of dermal vessels. The treatment is multipronged with anti-thrombotic measures such as anti-platelet drugs, systemic anticoagulants and fibrinolytic therapy taking precedence over anti-inflammatory agents. Colchicine, hydroxychloroquine, vasodilators, intravenous immunoglobulin, folic acid, immunosuppressive therapy and supportive measures are also of some benefit. A multidisciplinary approach would go a long way in the management of these patients resulting in relief from pain and physical as well as psychological scarring.Introduction
Livedoid vasculopathy is a hyalinising vascular disease characterised by thrombosis and ulceration of the lower extremities. Various terms are in use to designate livedoid vasculopathy including livedoid vasculitis, segmental hyalinizing vasculitis, atrophie blanche, livedo reticularis with ulcerations and painful purpuric ulcers with reticular pattern on the lower extremities. This entity was first described by Milian (1929) as atrophie blanche. Feldakar (1955) added the role of coagulation factors to the definition; the term “livedoid vasculopathy” was coined by Bard and Winkelmann (1967).[1],[2] The disorder continues to baffle researchers and clinicians alike by its enigmatic pathogenesis. The main mechnaism in the pathogenesis is hypercoagulability while inflammation plays a secondary role. Autoimmunity has recently been found to be contributory. It is a rare disorder with an estimated incidence of 1:100,000. There is female preponderance in the ratio of 3:1.[3] Disease manifestations start either in late adolescence or middle age with the mean age of onset at 32 years.
Pathogenesis
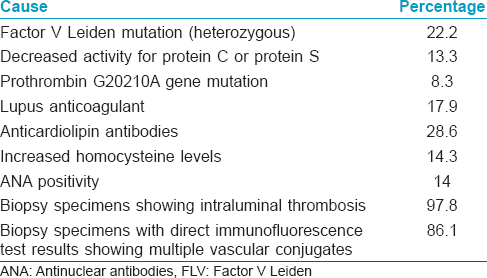
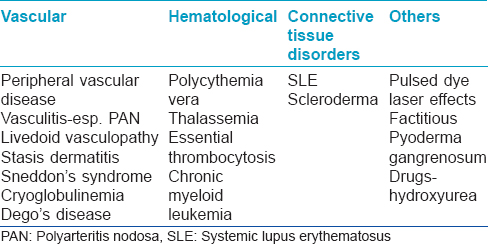
Initially livedoid vasculopathy was considered to be vasculitis; subsequently, its pathogenesis has been found to be related to occlusion of the cutaneous capillary microcirculation leading to thrombosis, ischemia and infarction. This explains the debilitating pain, paresthesia and hyperesthesia experienced by affected patients.[4] The thrombotic effect results from defects either in the endothelial cell plasminogen activation, platelet dysfunction or enhanced fibrin formation. Pericapillary deposition of fibrin and formation of thrombus act as a diffusion barrier impairing tissue oxygen supply causing ischemic infarction. Low tissue perfusion leads to poor wound healing. In sluggish circulation, there is ineffective killing of the microorganisms by leucocytes enhancing the chance of infection. A vicious cycle of tissue destruction, edema and thrombosis develops, further jeopardizing tissue perfusion. The important causative associations of livedoid vasculopathy are given in [Table - 1] and are discussed herewith.

The prothrombotic effect is substantiated by the presence of lupus anticoagulant and anticardiolipin antibodies in a significant number of patients, increased level of plasminogen activator inhibitor and low level of tissue plasminogen activator activity (<0.03IU/mL).[5] The prothrombotic state may also be augmented by some other factors;[6] these include increased serum homocysteine level, protein C and protein S deficiency, antithrombin III deficiency and abnormalities in fibrinolytic pathways. Some underlying diseases such as antiphospholipid antibody syndrome and sickle cell disease predispose to thrombotic episodes. Individuals with prothrombin G20210A gene mutation and factor V Leiden mutation are at high risk for thrombosis.
Hyperhomocysteinemia can be acquired or genetic. Acquired causes of homocysteinemia include nutritional deficiencies (folic acid, vitamin B12 and vitamin B6), renal failure, pernicious anemia, use of folic acid and vitamin B6 antagonists, cardiovascular and cerebral diseases and peripheral arterial occlusive disease. Genetic causes include defects in the enzymes cystathione-β-synthase, methylenetetrahydrofolate reductase and methionine synthase. The normal serum homocysteine concentration ranges between 5 to 15 μmol/L. Levels higher than this are associated with livedoid vasculopathy.[7],[8]
Activated protein C resistance is the more common inherited cause of thrombophilia associated with livedoid vasculopathy.[9],[10]
A glutamine with arginine substitution at position 506 of factor V (factor V Leiden mutation) causes protein C resistance. This mutation impairs activation of coagulation factor V by activated protein C leading to an increased risk of deep vein thrombosis. Heterozygous factor V Leiden mutation was found in 22.2% of patients with livedoid vasculopathy tested in a large American cohort.[11] The prevalence of heterozygous factor V Leiden mutation among ethnic Indians was found to be 10.7%.[12] Prothrombin gene mutation has an estimated prevalence of 0.7–2.6% in healthy people.[13] This mutation leads to increased plasma levels of prothrombin and several reports have associated these mutations with livedoid vasculopathy.[14],[15] Protein C deficiency has an estimated prevalence of <3%. The heterozygous deficiency of this protein with functional levels <65% is associated with an increased risk of thrombotic events which improves with antiplatelet treatment.[16],[17] Increased level of plasminogen activator inhibitor-1, a glycoprotein that inhibits the fibrinolytic system leads to inhibition of plasminogen and, therefore, venous thrombosis.[18]
The link between coagulation and inflammation is exemplified by the fact that protease-activated receptors-1 present on endothelium induces pro-inflammatory cytokine secretion (interleukin-6, interleukin-2, monocyte chemo-attractant protein-1) and adhesion molecule expression (intercellular adhesion molecule-1, P-selectin). This promotes leukocyte diapedesis and contributes to the inflammatory response.[19]
Autoimmunity may also be contributory. Therapeutic response to immunosuppressive and immunomodulatory agents and association with other autoimmune disorders support this fact. Increased perfusion pressure at the ankles seems to play an important role.
Rheumatoid arthritis, scleroderma, systemic lupus erythematosus, mixed and undifferentiated connective tissue diseases, polyarteritis nodosa and Sjogren's syndrome have been associated with livedoid vasculopathy. Patients who have antiphospholipid antibody syndrome with systemic lupus erythematosus are particularly predisposed. Presence of antiphospholipid antibodies, lupus anticoagulant and anticardiolipin antibodies in patients with livedoid vasculopathy also support this association.[20],[21]
Livedoid vasculopathy may also occur in patients with solid organ or hematological malignancies. Pregnancy may worsen livedoid vasculopathy, especially during the third trimester though there have been no reports of fetal compromise. A large proportion of cases, however, remain idiopathic.
An overview of the pathogenesis of livedoid vasculopathy is provided in [Figure - 1] and [Figure - 2]. The various etiopathological factors in livedoid vasculopathy are listed in [Table - 2].[12]
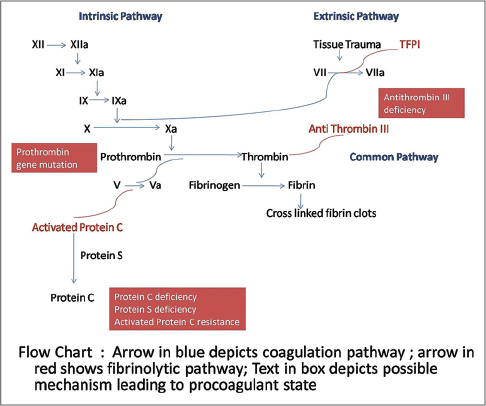 |
| Figure 1: Pathogenesis of thrombosis in livedoid vasculopathy, the coagulation and fibrinolytic pathways |
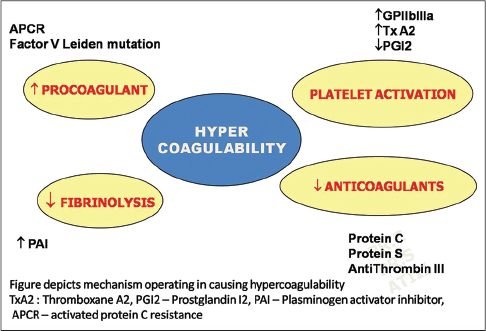 |
| Figure 2: Main thrombotic mechanisms in livedoid vasculopathy |
Clinical Features
Livedoid vasculopathy is characterized by a chronic, recurrent course with episodic exacerbations. The classical triad of manifestations is livedo reticularis, leg ulcerations and atrophie blanche.
The initial phase of livedo reticularis or livedo racemosa is characterized by livid, erythematous to purple net-like streaks, mainly due to abnormal perfusion of the cutaneous microcirculation. Initial lesions are purpuric macules, papules or ecchymotic streaks [Figure - 3]a and [Figure - 3]b distributed symmetrically on the dorsa of feet, ankles and lower legs. This is followed by the second stage characterized by acute-onset, painful ulcers of livedoid vasculopathy.
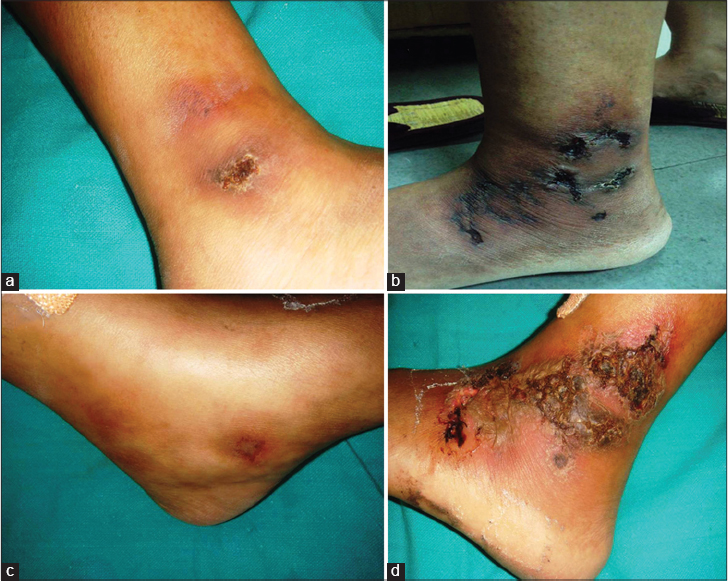 |
| Figure 3: Stages of ulcer formation in livedoid vasculopathy; (a) violaceous streaks just above the ankle with a solitary ulcer on the ankle. (b) Reticulate pattern of ecchymosis with impending ulceration. (c) Solitary impending ulcer on medial aspect of the foot. |
Inadequate blood supply due to thrombotic ischemia is the proposed cause for necrotic ulcers. These exquisitely painful ulcers are classically located asymmetrically on the ankle with extension to the dorsum and back of the foot, up to the distal leg [Figure - 3]c and [Figure - 3]d. Burning pain, severe enough to hamper daily activities, precedes the ulceration and is a clue for early diagnosis. Ulcers are characteristically small (4-6 mm), irregular, painful and recurrent. Rarely, these may coalesce to form big ulcers. Treatment at this stage may prevent ischemic cutaneous infarction and subsequent scarring. Ulcers may take 3-4 months to heal resulting in atrophie blanche (capillaritis alba). These are porcelain-white stellate scars usually surrounded by telangiectasias and hyperpigmentation representing irreversible infarction [Figure - 4]a and [Figure - 4]b. Atrophie blanche may also be seen in many other conditions [Table - 3].
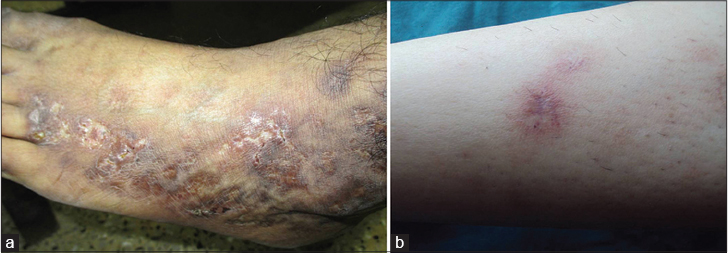 |
| Figure 4: Atrophie blanche: (a) scars on the entire lateral aspect of dorsum of the foot. (b) Characteristic telangiectasia around a healed scar |
Peripheral nervous system involvement may occur due to multifocal thrombosis and ischemia of the vasa nervorum. Systemic involvement is not a feature of idiopathic livedoid vasculopathy. However, when associated with autoimmune connective tissue disorders, multi-organ involvement may be present.
Diagnosis
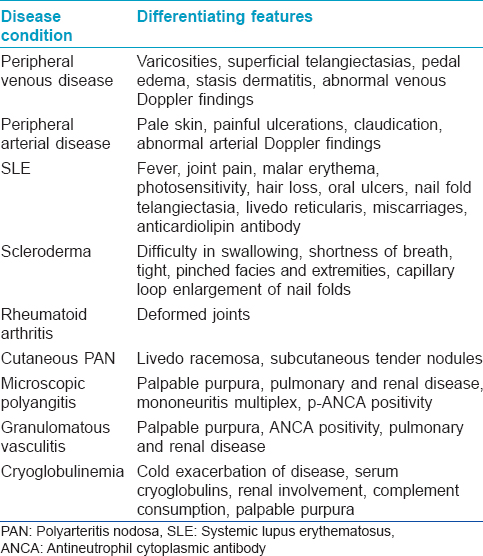
A detailed history, dermatological examination and laboratory work-up is essential to diagnose livedoid vasculopathy and its antecedent causes. Estimation of ankle brachial pressure index is important to rule out arterial and venous disorders. Investigations should be aimed at detecting associations and ruling out mimickers, as given in [Table - 4]. The clinical features to be taken into account in resolving the differential diagnosis have been presented in [Table - 5]. At times, it may be particularly challenging to differentiate it from polyarteritis nodosa. There is a higher prevalence of anti-myeloperoxidase antibodies and anti-phosphatidylserine prothrombin complex antibodies in the latter.[22] Polyarteritis nodosa is corticosteroid-responsive, while this drug is of little help in livedoid vasculopathy; on the other hand, anticoagulants improve livedoid vasculopathy.

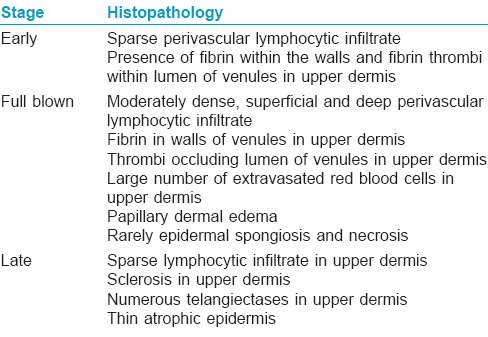
Histopathological examination of the ulcers is almost diagnostic. It is important to include the borders of the ulcer while doing a biopsy rather than taking only the base. There is characteristic fibrin occlusion and thrombus formation involving the upper and mid-dermal capillaries [Figure - 5]. As compared to primary vasculitis, there is hardly any perivascular inflammatory infiltrate. When present, the infiltrate is predominantly lymphocytic. Extravasation of red blood cells results from vessel wall damage and there is endothelial proliferation. Neutrophil infiltration and leukocytoclasia are usually absent (unlike in primary vasculitis). When neutrophils are found in the ulcerated area, it is usually secondary to the necrotizing ulcerative process. In the stage of atrophie blanche, there is hyalinization of the dermis and capillary walls. It is important to recognize that the histopathologic appearance varies at different stages of the disease [Table - 6].
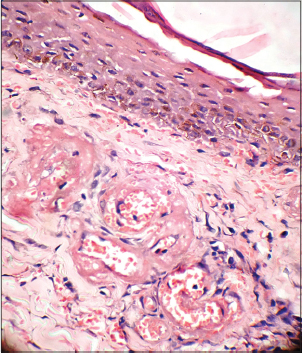 |
| Figure 5: Fibrin deposition and thrombus formation in the dermal vessels with absence of leukocytoclasia (H and E, ×400) |
Direct immunofluorescence study commonly demonstrates homogenous or granular deposition of immune complexes, fibrin and complement in the vessel wall. C3, IgM, IgA, IgG and fibrin, in descending order are the most common components of the deposits. The granular pattern of immunoreactant deposition, comprising mainly of C3 and IgM, is a differentiating feature from other immune complex deposition disorders.
Immune complexes may be deposited within thickened vessel walls indicating a secondary pathogenesis; most likely thrombosis is a primary event followed by fibrin deposition. It may also be a primary phenomenon with deposition of immune complexes leading to activation of the complement cascade in the microcirculation in turn causing activation of local coagulation. Electron microscopy shows dilated capillaries (diameter >100 μm) with thin endothelium and obliterated capillaries within a dense, fibrotic connective tissue matrix. Fibrin deposition and luminal occlusion of superficial blood vessels may be present. Erythrocytes and platelets are found trapped within the fibrin. Endothelial cells are replaced by fibrin in later stages.
Measurement of skin oxygen tension (transcutaneous oxygen pressure) or partial pressure of oxygen by transcutaneous oximetry adjacent to the ulcer can be used as a reliable marker of tissue ischemia and also to assess effects of therapy. The microcirculation can be studied better with Doppler flowmetry, laser Doppler perfusion imaging and microlymphography.
Treatment
Treatment of this condition is very challenging as there are no therapeutic guidelines due to low incidence and lack of multicenter studies. In cases with impending ulceration, prompt treatment reduces pain and prevents scarring. The main therapeutic options and treatment strategies have been presented in [Table - 7] and [Table - 8] and are discussed below.


Antiplatelet drugs
Aspirin is the most commonly used drug. It is a cyclooxygenase inhibitor that suppresses thromboxane A2 and prostaglandin I2 and thus promotes vasodilation, prevents thrombus formation and improves ulcer healing.[23],[24] It is usually used in combination with other anti-platelet agents in doses ranging from 75 to 325 mg, higher doses (up to 325 mg, thrice daily) offering better results. It may be helpful when there is associated sickle cell trait.[25] Dipyridamole also inhibits the synthesis of thromboxane A2 but stimulates the release of prostaglandin I2. The drug is used in a dose of 50 mg, thrice daily and may be combined with aspirin; fixed dose combinations are available. Pentoxifylline acts as a competitive non-selective phosphodiesterase inhibitor and thus reduces inflammation. It modifies red blood cell structure to reduce exocytosis, decreases blood viscosity and thus reduces platelet aggregation and thrombus formation.[26] Combination with other anti-platelet and anti-fibrinolytic agents enhances its effectiveness.[27],[28],[29] The recommended dose of pentoxyfylline is 400 mg thrice daily. Buflomedil hydrochloride, in addition to its anti-platelet effect, is a weak, non-specific calcium channel antagonist and alpha-blocker and is recommended orally at the dose of 150 mg, 3–4 times daily.[30] Sarpogrelate hydrochloride, an antagonist of 5-hydroxytryptamine 2A receptor (serotonin), has anti-platelet and vasodilator property and can be used at daily doses of 300 mg orally.[31]
Systemic anticoagulants
Heparin is used in refractory cases of livedoid vasculopathy as it not only inactivates the coagulation cascade but also decreases blood viscosity and increases fibrinolytic activity. A dose of 5000 U of heparin subcutaneously every 3 days may be enough to control the disease though 12 hourly doses have also been used.[32] The best evidence appears to be for low molecular weight heparin due to its effect on multiple stages of thrombin synthesis. Subcutaneous enoxaparin, 1 mg/kg up to twice daily has produced satisfactory results in many patients producing a rapid response with a favorable side effect profile.[33],[34] Dalteparin and nadroparin are newer alternatives to enoxaparin. Rivaroxaban, a new low molecular weight heparin is a direct inhibitor of factor Xa that was shown to be superior to enoxaparin in thrombo-prophylaxis and can prevent cutaneous ulcerations with the added advantage of oral administration.[35] Warfarin is another treatment option but requires to be monitored using international normalized ratio (to be maintained between 2 and 3) because of its effect on vitamin K-dependent factors. It may be helpful in patients with associated factor V Leiden mutation.[10],[36],[37] Other Vitamin K antagonists such as fluindione can also be used and also require monitoring of international normalized ratio which should be maintained between 2.5–3.5.[38] A visual analog scale for patient-assessment of pain can be used as an indicator for starting anticoagulant therapy. Patients must be counseled about the risk of bleeding when taking these drugs.
Fibrinolytic therapy
Livedoid vasculopathy is associated with elevated levels of plasminogen activator inhibitor which antagonizes the fibrinolytic pathway. Plasminogen activator inhibitors antagonize tissue plasminogen activator and urokinase-type plasminogen activator leading to a prothrombotic state. Fibrinolysis with recombinant tissue plasminogen activator lyses microvascular thrombi, restores the circulation and promotes wound healing.[5] Intravenous tissue plasminogen activator, is recommended in a dose of 10 mg administered intravenously initially 6 hourly and subsequently once daily for 14 days.[18],[39] Tissue plasminogen activator therapy may be considered especially in patients who have not responded to multiple conventional therapies. After tissue plasminogen activator therapy, maintenance with anti-platelet or anticoagulant agents has to be continued. Anabolic steroids such as stanozolol and danazol also have potential fibrinolytic activity.[40],[41] Danazol in low doses (200 mg/day or 4 mg/kg/day) for a short duration of 4–12 weeks has been found to be beneficial.[42],[43],[44] The adverse effects include hirsutism, breast atrophy, alopecia, weight gain, edema, hypertension, menstrual disturbances and clitoral hypertrophy.
Details of these antithrombotic drugs have been presented in [Table - 9].

Anti-inflammatory drugs
Colchicine is one of the primary anti-inflammatory drugs found to be beneficial in livedoid vasculopathy. Its anti-inflammatory effects are mediated through neutrophil inhibition. The drug is adminstered orally in doses of 0.5 mg, twice to thrice daily. Dapsone has a similar mechanism of action and is used in oral doses of 50–100 mg/day. Sulfasalazine, 1g 3 times daily orally has been found to improve ulcer healing. The drug exerts its anti-inflammatory effect through its sulfapyridine metabolite and probable inhibition of platelet aggregation by 5-aminosalicylic acid. It may also prevent cytokine release from mononuclear cells, thereby inhibiting platelet aggregation. Doxycycline at a dose of 100 mg twice a day can be used as adjuvant therapy.[45] Hydroxychloroquine, up to 0.6 mg/kg/day can also be used.[46]
Vasodilators
As vasospasm and peripheral vascular endothelial dysfunction are important etiological factors in livedoid vasculopathy, vasodilators such as nifedipine, cilostazol and nicotinic acid are reported to be beneficial as adjuvant therapy.[47]
Nutritional supplements
In cases with reduced methylenetetrahydrofolate reductase levels, additional supplementation of folic acid (5 mg/day), vitamin B6(1500 µg/day) and vitamin B12 is indicated.[8] Folic acid is required for homocysteine remethylation. Serum folate may be low due to dietary deficiency, heavy alcohol consumption, renal failure and in patients on anti-folate drugs such as methotrexate. Smoking also reduces serum folate concentration by interfering with its absorption. Individuals with methylenetetrahydrofolate reductase 677TT genotype, especially smokers, are recommended to increase folate intake to maintain adequate plasma homocysteine and serum folate levels.[48]
Immunosuppressive agents
Addition of immunosuppressive drugs to anti-platelet agents and anticoagulants has been found to enhance the efficacy of treatment. Prednisolone, azathioprine and colchicine are used to control disease activity.[49] The role of corticosteroids is controversial. In widespread disease, prednisolone has been used in many patients for rapid control of pain and early healing with beneficial results. In this context, the anti-inflammatory action of corticosteroids is probably of greater benefit than its immunosuppressive effect.[50] The drug may also act by its antifibrinolytic effect. Corticosteroids are used at the dosage of 0.5–1 mg/kg/day prednisolone equivalent. Cyclophosphamide, 1.5–2.5 mg/kg/day and azathioprine 2–3 mg/kg/day are other immunosuppressive agents that have been used for livedoid vasculopathy.[46]
Intravenous immunoglobulin
It acts by inhibiting Fc receptor function in macrophages, T cells and B cells leading to decreased cytokine production. There are various other proposed mechanisms of action that include reduction of immune complex deposition in small vessels, modulation of functional activity of T cells, solubilizing circulating and tissue-bound immune complexes, specific blockade of Fas via anti-Fas antibodies and inhibition of thromboxane synthetase thereby decreasing the vasoconstriction.[51],[52] When used in the treatment of livedoid vasculopathy, the combined anti-inflammatory and antithrombotic effects of intravenous immunoglobulin contribute to its effectiveness. It has been used as monthly infusions in the dose of 0.4–2g/kg.[53],[54]
Hyperbaric oxygen
In hyperbaric oxygen therapy, the person is allowed to breath 100% O2 under increased atmospheric pressure. This increased pressure enhances tissue oxygenation and microvascular perfusion by stimulating nitric oxide synthesis.[55] It also accelerates angiogenesis and fibrinolysis, inhibits collagen formation, accelerates fibroblast proliferation, diminishes tissue re-perfusion injury and has bacteriostatic and bactericidal effects. Growth of granulation tissue is accelerated leading to faster wound healing. Hyberbaric oxygen is administered for 1.5–2 hours, 1–3 times daily.[56],[57] Ulcers usually heal in 3–4 weeks making it a reasonably safe, fast and effective treatment option. Psoralen and ultraviolet A (PUVA) therapy may alter the ability of lymphocytes to respond to cytokine production. This therapy also induces release of immunosuppressive factors leading to decreased cell trafficking and altered proportion of lymphocyte subtypes in peripheral blood.[58],[59] In livedoid vasculopathy, PUVA has been used in an initial dose of ultraviolet A, 4 J/cm 2 increased by 0.5–1 J/cm 2 every week.[60]
Miscellaneous
Ketanserin, an S2 serotonergic receptor blocker, has been reported to be effective in the treatment of livedoid vasculopathy at a dose of 20 mg thrice daily. It prevents the vasoconstrictive effect of serotonin and thereby increases cutaneous blood flow.[61] Recently, alprostadil-alpha prostaglandin E1 has been found to be beneficial; the dose is 60 μg/day over 3 hours for 5 days, followed by a monthly infusion of 60 μg over 3 hours. Rituximab has also been used successfully.[62]
Preventive and supportive measures
Smoking is known to worsen morbidity in patients with livedoid vasculopathy.[58] Smoking significantly reduces peripheral blood flow, damages the vascular endothelium and increases the risk of venous thromboembolism.[63] In patients with livedoid vasculopathy, it will thus enhance tissue hypoxia and impair healing of ulcers. Smoking cessation is therefore an important preventive measure. Nine to 38% of patients with chronic venous insufficiency may suffer from livedoid vasculopathy. Compression therapy is helpful in the presence of a venous ulcer. It reduces edema and promotes ulcer healing. Avoidance of extreme changes in environmental temperature and topical application of perfusion-promoting formulations are some other preventive measures.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Feldaker M, Hines EA Jr, Kierland RR. Livedo reticularis with summer ulcerations. AMA Arch Derm 1955;72:31-42.
[Google Scholar]
|
| 2. |
Bard JW, Winkelmann RK. Livedo vasculitis. Segmental hyalinizing vasculitis of the dermis. Arch Dermatol 1967;96:489-99.
[Google Scholar]
|
| 3. |
Fritsch P, Zelger B. Livedo vasculitis. Hautarzt 1995;46:215-24.
[Google Scholar]
|
| 4. |
Amato L, Chiarini C, Berti S, Massi D, Fabbri P. Idiopathic atrophie blanche. Skinmed 2006;5:151-4.
[Google Scholar]
|
| 5. |
Klein KL, Pittelkow MR. Tissue plasminogen activator for treatment of livedoid vasculitis. Mayo Clin Proc 1992;67:923-33.
[Google Scholar]
|
| 6. |
Shankar S, Vasudevan B, Deb P, Langer V, Verma R, Nair V. Livedoid vasculopathy – A vasculitic mimic. Arthritis Rheum 2013;65:791.
[Google Scholar]
|
| 7. |
Gibson GE, Li H, Pittelkow MR. Homocysteinemia and livedoid vasculitis. J Am Acad Dermatol 1999;40(2 Pt 1):279-81.
[Google Scholar]
|
| 8. |
Meiss F, Marsch WC, Fischer M. Livedoid vasculopathy. The role of hyperhomocysteinemia and its simple therapeutic consequences. Eur J Dermatol 2006;16:159-62.
[Google Scholar]
|
| 9. |
Biedermann T, Flaig MJ, Sander CA. Livedoid vasculopathy in a patient with factor V mutation (Leiden). J Cutan Pathol 2000;27:410-2.
[Google Scholar]
|
| 10. |
Kavala M, Kocaturk E, Zindanci I, Turkoglu Z, Altintas S. A case of livedoid vasculopathy associated with factor V Leiden mutation: Successful treatment with oral warfarin. J Dermatolog Treat 2008;19:121-3.
[Google Scholar]
|
| 11. |
Hairston BR, Davis MD, Pittelkow MR, Ahmed I. Livedoid vasculopathy: Further evidence for procoagulant pathogenesis. Arch Dermatol 2006;142:1413-8.
[Google Scholar]
|
| 12. |
Pauer HU, Neesen J, Hinney B. Factor V Leiden and its relevance in patients with recurrent abortions. Am J Obstet Gynecol 1998; 178:129-130.
[Google Scholar]
|
| 13. |
Vicente V, González-Conejero R, Rivera J, Corral J. The prothrombin gene variant 20210A in venous and arterial thromboembolism. Haematologica 1999;84:356-62.
[Google Scholar]
|
| 14. |
Anavekar NS, Kelly R. Heterozygous prothrombin gene mutation associated with livedoid vasculopathy. Australas J Dermatol 2007;48:120-3.
[Google Scholar]
|
| 15. |
Gotlib J, Kohler S, Reicherter P, Oro AE, Zehnder JL. Heterozygous prothrombin G20210A gene mutation in a patient with livedoid vasculitis. Arch Dermatol 2003;139:1081-3.
[Google Scholar]
|
| 16. |
Boyvat A, Kundakçi N, Babikir MO, Gürgey E. Livedoid vasculopathy associated with heterozygous protein C deficiency. Br J Dermatol 2000;143:840-2.
[Google Scholar]
|
| 17. |
Baccard M, Vignon-Pennamen MD, Janier M, Scrobohaci ML, Dubertret L. Livedo vasculitis with protein C system deficiency. Arch Dermatol 1992;128:1410-1.
[Google Scholar]
|
| 18. |
Deng A, Gocke CD, Hess J, Heyman M, Paltiel M, Gaspari A. Livedoid vasculopathy associated with plasminogen activator inhibitor-1 promoter homozygosity (4G/4G) treated successfully with tissue plasminogen activator. Arch Dermatol 2006;142:1466-9.
[Google Scholar]
|
| 19. |
Papi M, Didona B, De Pità O, Frezzolini A, Di Giulio S, De Matteis W, et al. Livedo vasculopathy vs small vessel cutaneous vasculitis: Cytokine and platelet P-selectin studies. Arch Dermatol 1998;134:447-52.
[Google Scholar]
|
| 20. |
Di Giacomo TB, Hussein TP, Souza DG, Criado PR. Frequency of thrombophilia determinant factors in patients with livedoid vasculopathy and treatment with anticoagulant drugs – A prospective study. J Eur Acad Dermatol Venereol 2010;24:1340-6.
[Google Scholar]
|
| 21. |
Atsumi T, Ieko M, Bertolaccini ML, Ichikawa K, Tsutsumi A, Matsuura E, et al. Association of autoantibodies against the phosphatidylserine-prothrombin complex with manifestations of the antiphospholipid syndrome and with the presence of lupus anticoagulant. Arthritis Rheum 2000;43:1982-93.
[Google Scholar]
|
| 22. |
Kawakami T, Yamazaki M, Mizoguchi M, Soma Y. High titer of anti-phosphatidylserine-prothrombin complex antibodies in patients with cutaneous polyarteritis nodosa. Arthritis Rheum 2007;57:1507-13.
[Google Scholar]
|
| 23. |
Yang LJ, Chan HL, Chen SY, Kuan YZ, Chen MJ, Wang CN, et al. Atrophie blanche. A clinicopathological study of 27 patients. Changgeng Yi Xue Za Zhi 1991;14:237-45.
[Google Scholar]
|
| 24. |
Ibbotson SH, Layton AM, Davies JA, Goodfield MJ. The effect of aspirin on haemostatic activity in the treatment of chronic venous leg ulceration. Br J Dermatol 1995;132:422-6.
[Google Scholar]
|
| 25. |
El Khoury J, Taher A, Kurban M, Kibbi AG, Abbas O. Livedoid vasculopathy associated with sickle cell trait: Significant improvement on aspirin treatment. Int Wound J 2012;9:344-7.
[Google Scholar]
|
| 26. |
Sams WM Jr. Livedo vasculitis. Therapy with pentoxifylline. Arch Dermatol 1988;124:684-7.
[Google Scholar]
|
| 27. |
Sauer GC. Pentoxifylline (Trental) therapy for the vasculitis of atrophie blanche. Arch Dermatol 1986;122:380-1.
[Google Scholar]
|
| 28. |
Ely H, Bard JW. Therapy of livedo vasculitis with pentoxifylline. Cutis 1988;42:448-53.
[Google Scholar]
|
| 29. |
Zecevic RD. Livedo vasculitis. Vojnosanit Pregl 2001;58:263-6.
[Google Scholar]
|
| 30. |
Drucker CR, Duncan WC. Antiplatelet therapy in atrophie blanche and livedo vasculitis. J Am Acad Dermatol 1982;7:359-63.
[Google Scholar]
|
| 31. |
Antunes J, Filipe P, André M, Fraga A, Miltenyi G, Marques Gomes M. Livedoid vasculopathy associated with plasminogen activator inhibitor-1 promoter homozygosity (4G/4G) and prothrombin G20210A heterozygosity: Response to t-PA therapy. Acta Derm Venereol 2010;90:91-2.
[Google Scholar]
|
| 32. |
Jetton RL, Lazarus GS. Minidose heparin therapy for vasculitis of atrophie blanche. J Am Acad Dermatol 1983;8:23-6.
[Google Scholar]
|
| 33. |
Guinier MC, Gauthier O, Chatelan M, Lemoing M, Boisseau MR. Low-molecular-weight heparin in leg ulcers due to microangiopathy Results with curative doses in nineteen patients. Semaine Des Hopitaux 1996;72:231-6.
[Google Scholar]
|
| 34. |
Hairston BR, Davis MD, Gibson LE, Drage LA. Treatment of livedoid vasculopathy with low-molecular-weight heparin: Report of 2 cases. Arch Dermatol 2003;139:987-90.
[Google Scholar]
|
| 35. |
Kerk N, Drabik A, Luger TA, Schneider SW, Goerge T. Rivaroxaban prevents painful cutaneous infarctions in livedoid vasculopathy. Br J Dermatol 2013;168:898-9.
[Google Scholar]
|
| 36. |
Browning CE, Callen JP. Warfarin therapy for livedoid vasculopathy associated with cryofibrinogenemia and hyperhomocysteinemia. Arch Dermatol 2006;142:75-8.
[Google Scholar]
|
| 37. |
Davis MD, Wysokinski WE. Ulcerations caused by livedoid vasculopathy associated with a prothrombotic state: Response to warfarin. J Am Acad Dermatol 2008;58:512-5.
[Google Scholar]
|
| 38. |
Francès C, Barete S. Difficult management of livedoid vasculopathy. Arch Dermatol 2004;140:1011.
[Google Scholar]
|
| 39. |
Agirbasli M, Eren M, Eren F, Murphy SB, Serdar ZA, Seckin D, et al. Enhanced functional stability of plasminogen activator inhibitor-1 in patients with livedoid vasculopathy. J Thromb Thrombolysis 2011;32:59-63.
[Google Scholar]
|
| 40. |
Rizzo SC, Grignani G, Gamba G, Nalli G. Fibrinolysis induced by danazol. Blut 1986;53:351-2.
[Google Scholar]
|
| 41. |
Shigekiyo T, Tomonari A, Uno Y, Kishi Y. Danazol therapy in hypoplasminogenemia. Thromb Haemost 1992;68:233-4.
[Google Scholar]
|
| 42. |
Hsiao GH, Chiu HC. Livedoid vasculitis. Response to low-dose danazol. Arch Dermatol 1996;132:749-51.
[Google Scholar]
|
| 43. |
Hsiao GH, Chiu HC. Low-dose danazol in the treatment of livedoid vasculitis. Dermatology 1997;194:251-5.
[Google Scholar]
|
| 44. |
Wakelin SH, Ellis JP, Black MM. Livedoid vasculitis with anticardiolipin antibodies: Improvement with danazol. Br J Dermatol 1998;139:935-7.
[Google Scholar]
|
| 45. |
Keller MS, Lee J, Webster GF. Livedoid thrombotic vasculopathy responding to doxycycline therapy. J Clin Aesthet Dermatol 2008;1:22-4.
[Google Scholar]
|
| 46. |
Gan EY, Tang MB, Tan SH, Chua SH, Tan AW. A ten-year retrospective study on livedo vasculopathy in Asian patients. Ann Acad Med Singapore 2012;41:400-6.
[Google Scholar]
|
| 47. |
Purcell SM, Hayes TJ. Nifedipine treatment of idiopathic atrophie blanche. J Am Acad Dermatol 1986;14 (5 Pt 1):851-4
[Google Scholar]
|
| 48. |
Anderson CA, Beresford SA, McLerran D, Lampe JW, Deeb S, Feng Z, Motulsky AG. Response of serum and red blood cell folate concentrations to folic acid supplementation depends on methylenetetrahydrofolate reductase C677T genotype: Results from a crossover trial. Mol Nutr Food Res 2013;57:637-44.
[Google Scholar]
|
| 49. |
Lee SS, Ang P, Tan SH. Clinical profile and treatment outcome of livedoid vasculitis: A case series. Ann Acad Med Singapore 2003;32:835-9.
[Google Scholar]
|
| 50. |
Marzano AV, Vanotti M, Alessi E. Widespread livedoid vasculopathy. Acta Derm Venereol 2003;83:457-60.
[Google Scholar]
|
| 51. |
Ravat FE, Evans AV, Russell-Jones R. Response of livedoid vasculitis to intravenous immunoglobulin. Br J Dermatol 2002;147:166-9.
[Google Scholar]
|
| 52. |
Oravec S, Ronda N, Carayon A, Milliez J, Kazatchkine MD, Hornych A. Normal human polyspecific immunoglobulin G (intravenous immunoglobulin) modulates endothelial cell function in vitro. Nephrol Dial Transplant 1995;10:796-800.
[Google Scholar]
|
| 53. |
Amital H, Levy Y, Shoenfeld Y. Use of intravenous immunoglobulin in livedo vasculitis. Clin Exp Rheumatol 2000;18:404-6.
[Google Scholar]
|
| 54. |
Kreuter A, Gambichler T, Breuckmann F, Bechara FG, Rotterdam S, Stücker M, et al. Pulsed intravenous immunoglobulin therapy in livedoid vasculitis: An open trial evaluating 9 consecutive patients. J Am Acad Dermatol 2004;51:574-9.
[Google Scholar]
|
| 55. |
Fernandes TD. Hyperbaric medicine. Acta Med Port 2009;22:323-34.
[Google Scholar]
|
| 56. |
Yang CH, Ho HC, Chan YS, Liou LB, Hong HS, Yang LC. Intractable livedoid vasculopathy successfully treated with hyperbaric oxygen. Br J Dermatol 2003;149:647-52.
[Google Scholar]
|
| 57. |
Juan WH, Chan YS, Lee JC, Yang LC, Hong HS, Yang CH. Livedoid vasculopathy: Long-term follow-up results following hyperbaric oxygen therapy. Br J Dermatol 2006;154:251-5.
[Google Scholar]
|
| 58. |
Choi HJ, Hann SK. Livedo reticularis and livedoid vasculitis responding to PUVA therapy. J Am Acad Dermatol 1999;40 (2 Pt 1):204-7.
[Google Scholar]
|
| 59. |
Lee JH, Choi HJ, Kim SM, Hann SK, Park YK. Livedoid vasculitis responding to PUVA therapy. Int J Dermatol 2001;40:153-7.
[Google Scholar]
|
| 60. |
Tuchinda C, Leenutaphong V, Sudtim S, Lim HW. Refractory livedoid vasculitis responding to PUVA: A report of four cases. Photodermatol Photoimmunol Photomed 2005;21:154-6.
[Google Scholar]
|
| 61. |
Rustin MH, Bunker CB, Dowd PM. Chronic leg ulceration with livedoid vasculitis, and response to oral ketanserin. Br J Dermatol 1989;120:101-5.
[Google Scholar]
|
| 62. |
Zeni P, Finger E, Scheinberg MA. Successful use of rituximab in a patient with recalcitrant livedoid vasculopathy. Ann Rheum Dis 2008;67:1055-6.
[Google Scholar]
|
| 63. |
Hansson PO, Eriksson H, Welin L, Svärdsudd K, Wilhelmsen L. Smoking and abdominal obesity: Risk factors for venous thromboembolism among middle-aged men: “The study of men born in 1913”. Arch Intern Med 1999;159:1886-90.
[Google Scholar]
|
Fulltext Views
16,607
PDF downloads
4,699





![[Table - 1]](#tbl_ijdvl_2016_82_5_478_183635_t6.jpg){kind=link}
![[Figure - 1]](#fig_ijdvl_2016_82_5_478_183635_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2016_82_5_478_183635_f2.jpg){kind=link}
![[Table - 2]](#tbl_ijdvl_2016_82_5_478_183635_t7.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2016_82_5_478_183635_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2016_82_5_478_183635_f4.jpg){kind=link}
![[Table - 3]](#tbl_ijdvl_2016_82_5_478_183635_t8.jpg){kind=link}
![[Table - 4]](#tbl_ijdvl_2016_82_5_478_183635_t9.jpg){kind=link}
![[Table - 5]](#tbl_ijdvl_2016_82_5_478_183635_t10.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2016_82_5_478_183635_f5.jpg){kind=link}
![[Table - 6]](#tbl_ijdvl_2016_82_5_478_183635_t11.jpg){kind=link}
![[Table - 7]](#tbl_ijdvl_2016_82_5_478_183635_t12.jpg){kind=link}
![[Table - 8]](#tbl_ijdvl_2016_82_5_478_183635_t13.jpg){kind=link}
![[Table - 9]](#tbl_ijdvl_2016_82_5_478_183635_t14.jpg){kind=link}