Translate this page into:
Melanocyte spheroids are formed by repetitive long-term trypsinization
2 Department of Dermatology, The First Affiliated Hospital of Bengbu Medical College, Anhui, China
Correspondence Address:
Ru-Zhi Zhang
Department of Dermatology, The Third Affiliated Hospital of Soochow University, 185 Juqian Road, Changzhou - 213003
China
| How to cite this article: Li D, Zhang RZ, Shi HX, Yang YH, Tian T, Wang L. Melanocyte spheroids are formed by repetitive long-term trypsinization. Indian J Dermatol Venereol Leprol 2019;85:258-265 |
Abstract
Background: Autologous melanocyte transplantation plays an important role in the treatment of vitiligo.
Objective: Previous studies have indicated that, compared with melanocytes growing in monolayers, melanocyte spheroids have a better survival in growth factor- and serum-deprived conditions.
Methods: Melanocyte spheroids were obtained from human epidermis by repetitive long-term trypsinization and maintained an aggregated morphology for a short period in certain conditions.
Results: Melanocyte spheroids were capable of growing into normal dendritic melanocytes in monolayer when they were harvested and reinoculated in 24-well plates. Immunohistochemical analysis of the melanocyte spheroids revealed that they were positive for HMB45, a melanosome-specific marker. No melanomas occurred when melanocyte spheroids were transplanted into mice.
Conclusion: Our study provides a promising approach for melanocyte transplantation to treat vitiligo.
Introduction
Vitiligo is an acquired dermatological disorder characterized by circumscribed depigmented macules due to the loss of functional melanocytes in the epidermis.[1],[2] Therapeutic options for vitiligo include the application of topical steroids, psoralen plus ultraviolet A (PUVA), narrowband ultraviolet B, biologics, immunosuppressants, low energy laser irradiation and surgical therapy.[3],[4] In intractable cases of vitiligo, autologous melanocyte transplantation may be used to repigment the skin, wherethe vitality and purification of the transplanted melanocytes are crucial for effective results.
In the process of cell transplantation, providing a ''survival advantage''to cells is very important. It has been found that melanocytes spontaneously grow into three-dimensional spheroids on a chitosan-coated surface in growth factor- and serum-deprived conditions, and preculturing melanocytes into spheroids can provide them a survival advantage.[5] Lin et al. reported that, depending on the seeding density and culture time, melanocytes can assume a monolayer or a spheroid morphology.[6]
Melanocytes in human skin are roughly divided into two distinct populations: mature differentiated cells and melanocyte stem cells (MSCs). Melanocyte stem cells and/or their immediate progenies are slow-cycling and self-renewing, and generally stress-tolerant. In 2013, Shigemoto proposed a novel approach to collect satellite cells from skeletal muscles based on their stress tolerance. They suggested that long-term trypsinization is a safe and inexpensive method to efficiently collect tissue stem cells.[7]
The aim of this study was to observe the changes of primary melanocytes in culture treated by repetitive long-term trypsinization and to investigate the biological functions of melanocyte spheroids.
Methods
All procedures and human experiments were approved by the Ethics Committee of theFirst People's Hospital of Changzhou (Changzhou, China) [permit number (2013) number 4].
Preparation of adult human epidermal melanocytes
Primary cultured cells were harvested from adult foreskin by digesting it with dispase and trypsin sequentially. These cells were cultured in M254 Medium supplemented with human melanocyte growth supplement, which is suitable for melanocyte growth but not for other types of cells.[18],[19],[20]
Skin specimens were obtained from adult circumcisions and were immediately immersed in iodophor solution for 5 min, then washed with phosphate-buffered saline supplemented with 400 U/mL penicillin and 400 μg/mL streptomycin. Subcutaneous adipose tissues were removed manually using eye scissors. The remaining tissues, including the epidermis and dermis, were washed again with phosphate-buffered saline and cut into small pieces (approximately 0.5 cm × 1 cm). The epidermis was separated from the dermis after treatment with 0.25% dispase (cat. no. 4942078001, Roche, Basel, SUI) for 2 h. The epidermal sheets obtained were treated with 0.25% trypsin- ethylenediaminetetraacetic acid (cat. no. 25200-056, Gibco, Grand Island, NY, USA) for 15 min to produce a cellular suspension which was filtered through a 200-μm filter and then centrifuged at 1000 rpm for 3 min.[8] The cells obtained were resuspended and maintained in a growth medium consisting of 254 Medium (cat.no. M-254-500, Gibco, NY, USA) containing human melanocyte growth supplement-2 (cat.no.S-016-5, Gibco, NY, USA), in 5% CO2 at 37°C. Culture media were changed every 3 days.[9],[10]
Repeated long-term trypsinization
When melanocytes in primary culture reached 70–80% confluence, the medium was discarded, adherent melanocytes were gently rinsed with 0.01 M phosphate-buffered saline and then incubated with 0.25% trypsin-ethylenediaminetetraacetic acid (EDTA) solution for 2 h. Subsequently, the cells were washed with 0.01 M phosphate-buffered saline and suspended in a 5 mL Falcon tube which was vortexed for 1 min at 1,800 rpm and then centrifuged at 400 rpm for 10 min. Finally, the supernatant containing any dead cells was removed. The surviving cells were resuspended in 254 Medium and were then reseeded in 24-well plates. Approximately 1 week later, when cells in the second passage were 70–80% confluent, the above processes of trypsinization and reseeding were performed, but with the incubation time being 4 h. A similar sequence was again carried out when the third passage cells became 80–90% confluent, with an incubation time of 6 h.[11]
Morphological observations and immunofluorescence staining
For morphological observations, cells and spheroids adhering to the walls of the dishes were photographed using an inverted phase contrast microscope. Melanocyte spheroids were picked up using the blunt point of a transfer pipette. Actually, with a smooth surface they could be detached intact by gently pipetting the culture medium. They were then reseeded in another 24-well culture plate. When these spheroids attached to the bottom of the plate and grew outwards, staining could be carried out. The cells were fixed in 4% paraformaldehyde for 15 min at room temperature. Then they were incubated in 0.3% Triton X-100 in phosphate-buffered saline for 10 min. After two successive washes with phosphate-buffered saline, the cells were incubated with a blocking solution containing 5% bovine serum albumin (cat.no. 10099141, Gibco) at room temperature for 60 min. They were subsequently stained with HMB45 antibody (1:50; cat.no.M0634, Dako), which was diluted in 1% bovine serum albuminin phosphate-buffered saline and then incubated overnight at 4°C. After being washed three times in phosphate-buffered saline, these cells were incubated with a secondary antibody (1:400; cat.no.ab150105, Abcam) in 1% bovine serum albumin in phosphate-buffered saline for 2 h at room temperature in the dark. An isotype-matched donkey anti-mouse IgG (1:400;cat.no.ab150105, Abcam) was used as a negative control.[12] The results were observed using a fluorescence microscope (Olympus, Tokyo, Japan).[13]
Electron microscopy
Melanocyte spheroids were collected and fixed in 2.5% glutaraldehyde (buffered to pH 7.2 with sodium cacodylate) for 4 h. Then they were post-fixed in 1% (wt/vol) osmium tetroxide for 1 h. The specimens were dehydrated sequentially using ethanol at concentrations of 30, 50, 70, 80, 90 and 100%, each phase for 10 min. The specimens were then embedded in Epon-Araldite and cut into ultrathin sections (70 nm thick) using a Leica Ultracut UCT ultramicrotome (EM UC6, Leica, Wetzlar, Germany), mounted on copper grids and stained with lead and uranyl solutions.[14],[15] Finally, these sections were observed using a transmission electron microscope (JEM-2100, JEOL, Tokyo, Japan).
Transplantation
To check if the cells after repeated trypsinization might become melanoma cells, we inoculated them subcutaneously into nude mice.[16] Subcutaneous transplantation is easy to perform and allows simple monitoring of tumor development. Prior to inoculation, melanocyte spheroids were dissociated into single-cell suspensions to enable the transplantation of approximately 1.5 × 10[5] cells. To increase sensitivity, the transplanted cells were mixed with Matrigel. We monitored the transplanted animals for the development of tumors for a period of 4 months.
Immunohistochemical examination of tissues
After 4 months, the skin tissue of mice at each transplanted site was removed, fixed, embedded and cut into sections for immunohistochemical examination.[17] After deparaffinization and hydration, these sections were subjected to blocking of endogenousperoxidase activity, and were then treated with 10 mMEDTA at pH 6.0, at 100°C in a steamer to allow antigen retrieval. Immunohistochemical investigations were performed using the HMB45 antibody (1:50; cat.no. M0634, Dako) diluted in 1% bovine serum albuminin phosphate-buffered saline at pH 7.6. The slides were incubated with the primary antibody described above for 2 h at 37°C. Antigen-antibody binding was detected by means of the EnVision peroxidase system (code K1491; Dako). Staining was achieved using 3,3′-diaminobenzidine (Dako) diluted in 3% H2O2 in phosphate-buffered saline at pH 7.6. The same specimens were used as negative controls by omitting the primary antibody and replacing it with 1% bovine serum albumin.
Results
Primary cultures
When reaching 70–80% confluence, the cultured cells comprised a majority of melanocytes, a small number of keratinocytes and a few fibroblasts [Figure - 1].The melanocytes had small bodies with a good diopter, and multiple dendrites were obvious; however, they did not show clonal growth behavior. In contrast, the keratinocytes showed colony-like growth and were arranged densely with a clear boundary.
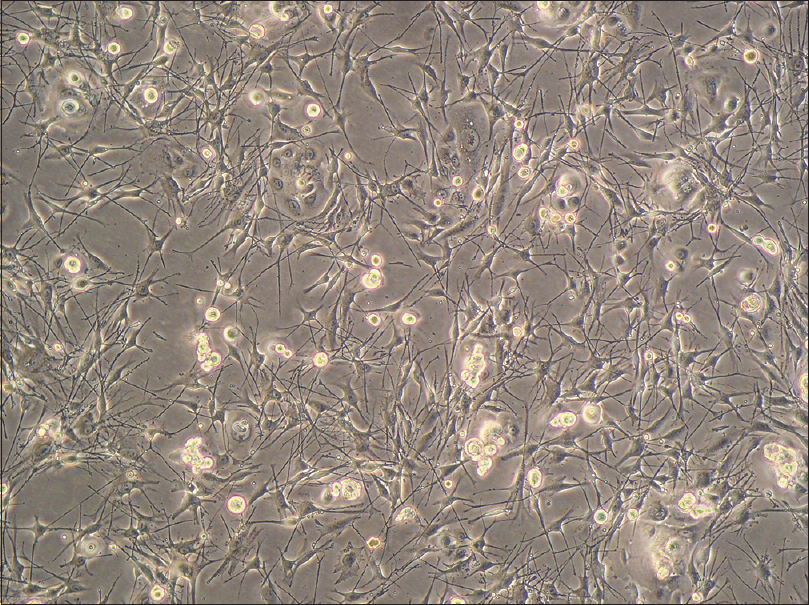 |
| Figure 1: Primary culture. The cultured cells comprised a majority of melanocytes, a small number of keratinocytes and a few fibroblasts (inverted phase contrast microscopy, ×100) |
Enrichment of melanocytes after the first long-term trypsinization
In preliminary experiments, we subjected primary cultured cells to long-term trypsinization for 0.5, 1, 2, 3, 4, 5, 6 and 8 h, to determine the best conditions for high survival ratio of melanocytes and the elimination of keratinocytes and fibroblasts. The results indicated that long-term trypsinization for 2 h was the most suitable condition for the primary culture. In that case, the proportion of melanocyte stem cells increased, and keratinocytes and fibroblasts almost disappeared, although we do not know the underlying mechanisms.
The surviving cells grew quickly and achieved 70–80% confluence on the seventh day after long-term trypsinization. At this time, the cells displayed the classic form of melanocytes, with each cell having multiple very long dendrites [Figure - 2]a. A long-term trypsinization of 4 h was performed on these cells. Four days later, the cells achieved 70–80% confluence and tended to form clusters [Figure - 2]b. Larger compact cellular melanocyte spheroids were observed on the third day after yet another long-term trypsinization of 6 h [Figure - 2]c and [Figure - 2]d. Between the melanocyte spheroids, dendritic melanocytes were still present.
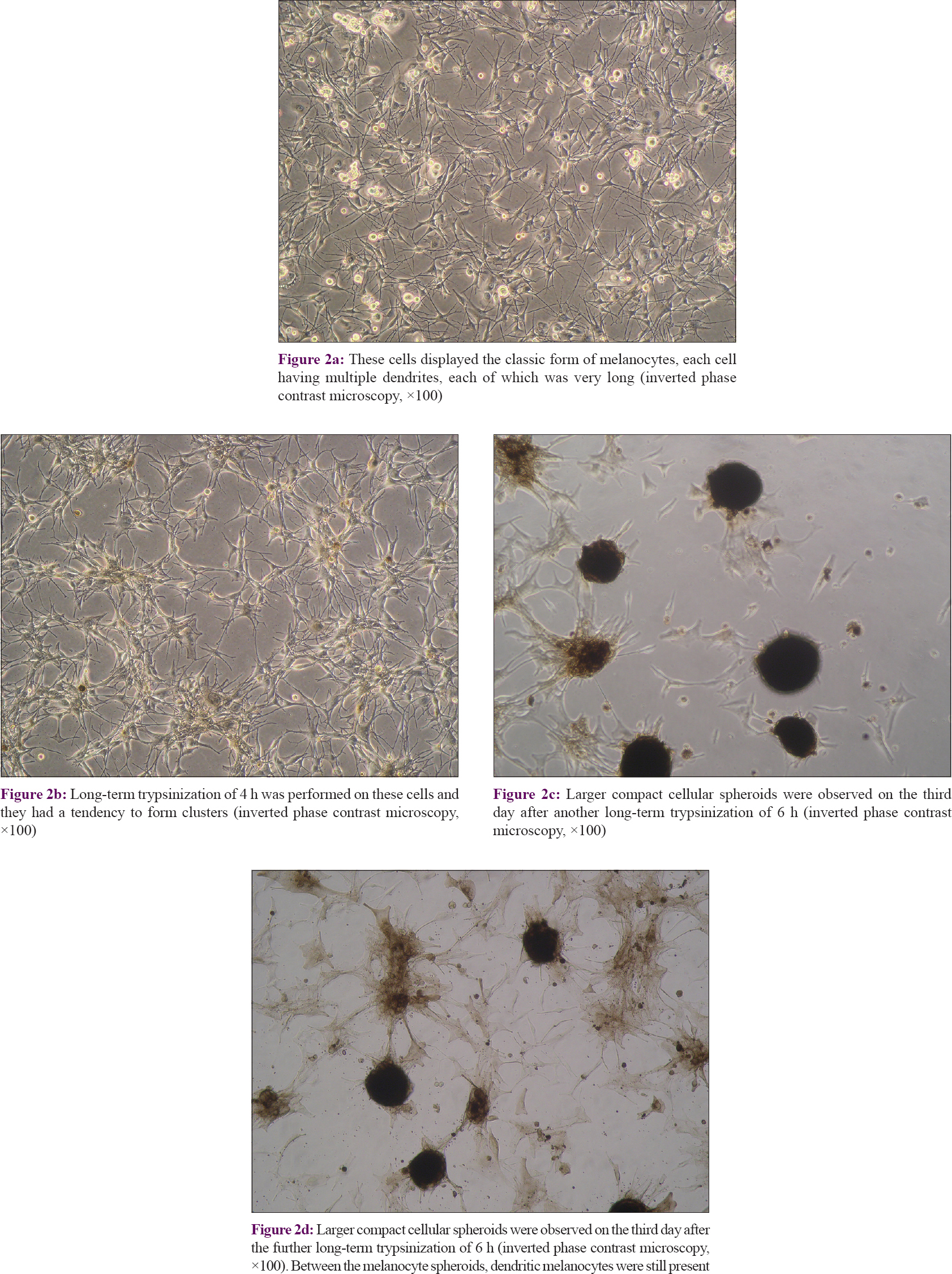 |
| Figure 2: |
Culture of cellular spheroids
The diameters of melanocyte spheroids ranged from 50–250 μm and they were capable of growing into monolayer dendritic melanocytes when reseeded in culture plates [Figure - 3]a,[Figure - 3]b, [Figure - 3]c. Over time, these melanocyte spheroids gradually disintegrated and were replaced by a monolayer of cells that grew vigorously. After passage, few melanocyte spheroids could be seen [Figure - 3]d. The second-generation cells showed a short spindle-shape, with good diopters and bipolar protrusions, characteristic of immature melanocytes.
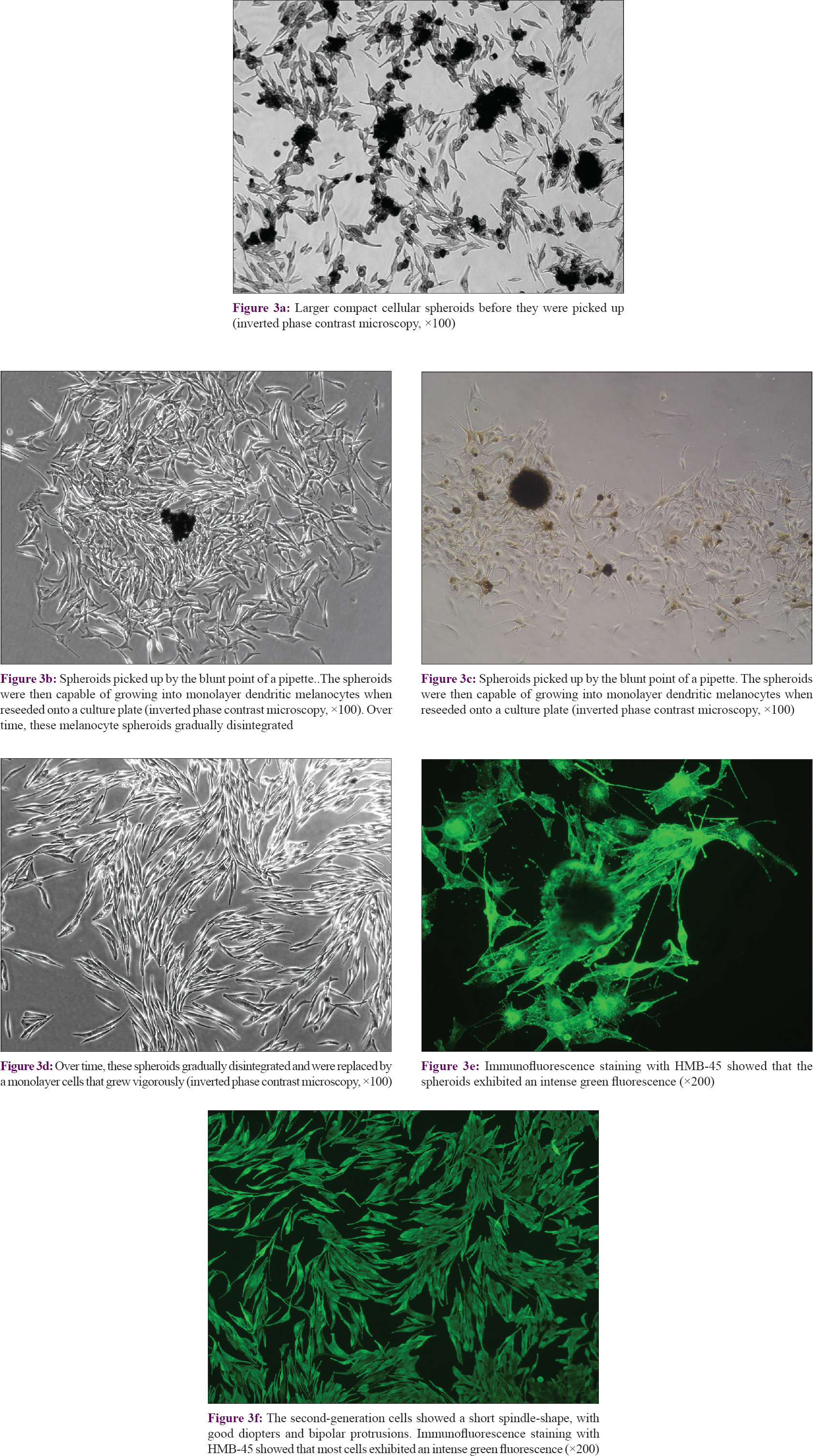 |
| Figure 3: |
On immunofluorescence staining with HMB45, most cells exhibited an intense green fluorescence [Figure - 3]e and [Figure - 3]f, indicating the presence of moderate to high levels of HMB45 antigen, which is a specific marker for melanosomes.[21] Fluorescence was stronger at the tips of the cellular dendrites and at the periphery of the cell bodies, demonstrating that those regions were rich in melanosomes. The staining pattern confirmed that these cells were indeed melanocytes, and further demonstrated that the melanocyte spheroids were viable.
Electron microscopy
Transmission electron microscopy revealed that the nucleus/cytoplasm ratio of these cells was high and chromatin aggregation in the nuclei was poor [Figure - 4]a. The dense melanin granules appeared as uneven masses [Figure - 4]b. Double-membrane-walled structures containing electron-dense material were found, typical for melanin-containing autophagosomes [Figure - 4]c.[22] A few melanin-containing autophagosomes had exocytosed into the intercellular spaces [Figure - 4]d. These findings indicated that autophagy may have occurred in melanocytes under conditions of stress.
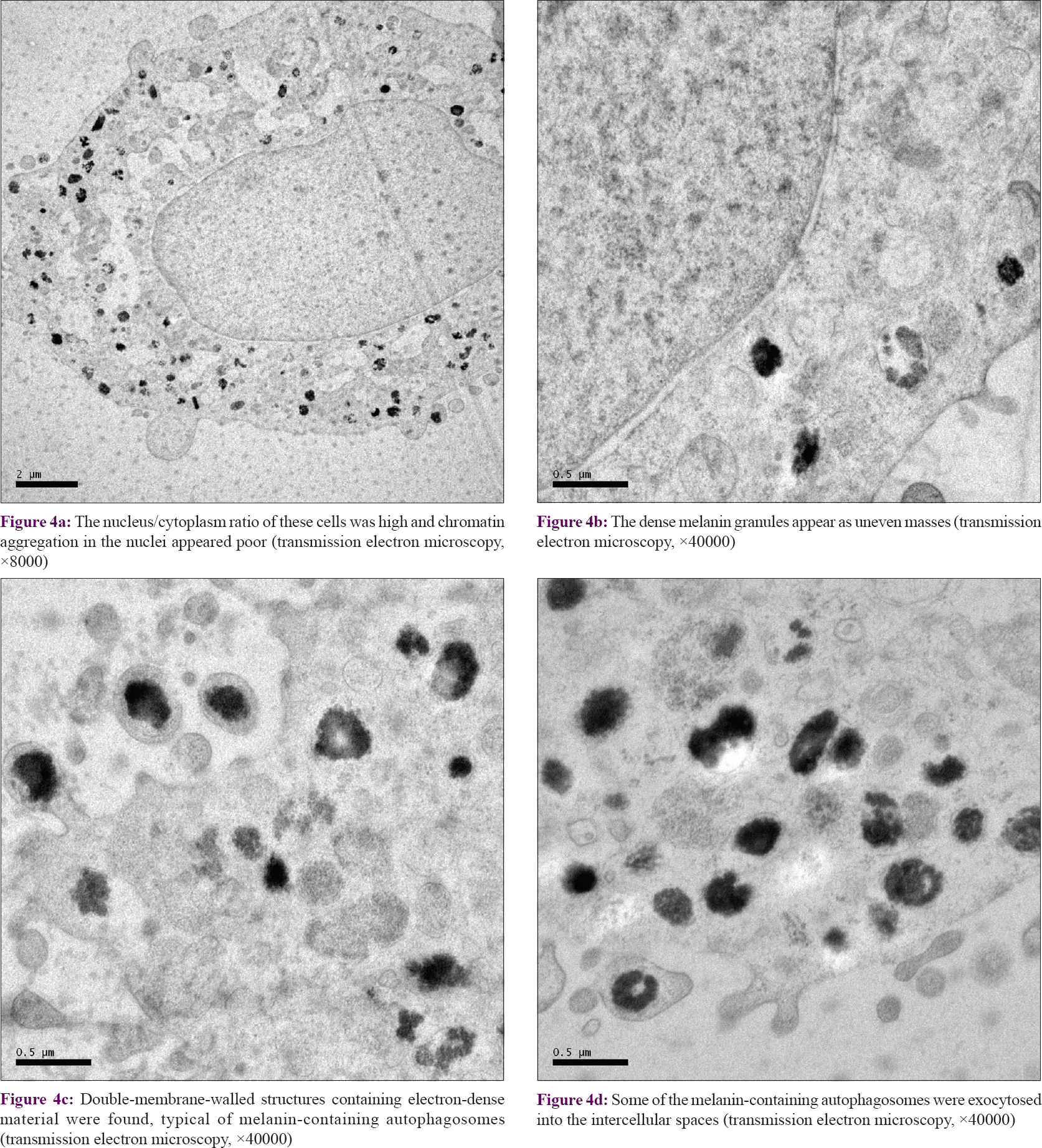 |
| Figure 4: |
Transplantation and immunohistochemical examination of tissues
30 μl of melanocyte suspension (approximately 1.5 × 10[5] cells) was slowly injected into the subcutaneous tissue of the right rear leg of each mouse. We observed the skin for changes on a daily basis, and found a black spot at the injection site on the sixth day. Four months later, punch biopsies were taken. Hematoxylin and eosin staining of the excised tissue was normal. Immunohistochemical staining revealed many HMB45-positive cells located in the deep dermis and subcutaneous tissue [Figure - 5], but there was no evidence of melanoma tissue.
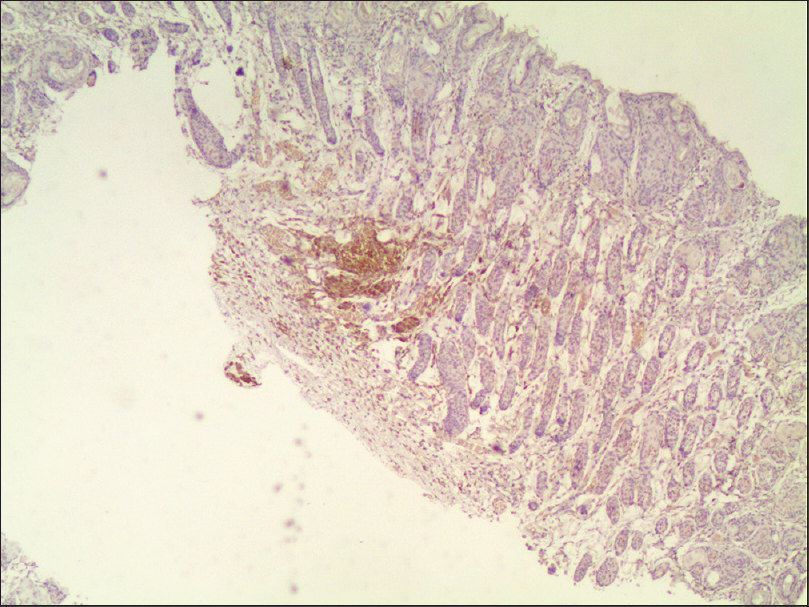 |
| Figure 5: Immunohistochemical staining revealed many HMB45-positive cells located in the deep dermis and subcutaneous tissue (light microscopy, ×40) |
Discussion
Vitiligo is an acquired depigmenting disorder with great cosmetic importance. Various treatment modalities have been tried to repigment vitiliginous skin.[23] Melanocyte transplantationis a cellular grafting procedure that aims to replenish the lost melanocytes inthe vitiliginous epidermis. In general, epidermal melanocytes are mature cells showing multiple dendrites and pigmentation. Melanocyte stem cells and/or their immediate descendants have a potential to provide a sufficient supply of melanocytes for cell-based treatments due to their high proliferative capacity, but the separation and purification of these cells is still a challenging problem.
Flow cytometry is widely used to collect stem cells from tissues by detecting specific surface markers, but it cannot be directly used for the collection of some tissue stem cells with no known specific markers, such as melanocyte stem cells. Some authors have developed a very efficient and inexpensive method to collect satellite cells.[24] They reported that a long-term trypsinization of 6 h was the most effective method to enrich mouse satellite cells and 3 h for human cells, due to species-specific differences. We tried long-term trypsinization for varying periods, and found that lengthening the time did less damage to cell activity. Shigemoto et al. also described that the ratio of stem cells among primary cultured cells was substantially increased by long-term trypsinization. This was also verified in our experiments, considering the time required to reach cell confluence. It took about 7–8 days to reach 70–80% cell fusion after the first long-term trypsinization, but the second and third periods were approximately 3–4 days. As the proportion of melanocyte stem cells gradually increased, the total number of cells increased rapidly. In other words, the repeated trypsinization greatly increased the proportion of melanocyte stem cells.
In previous experiments, trypsinization took only 3 min at 37°C, in the third generation of melanocytes mixed with a large number of fibroblasts. In our experiments, we subjected primary cultured cells to long-term trypsinization for 2, 4 and 6 h. The activity and purity of obtained melanocytes by our method were higher. Furthermore, it requires no expensive devices and can be easily performed in any laboratory. In the transplantation experiment, we confirmed that although these cells underwent repeated long-term trypsinization, they did not become melanoma cells. Though these findings suggest that it does not significantly affect the properties of the original cells, they are insufficient to recommend using these melanocytes immediately in the clinic, and genetic testing may be needed.
It is known that neuron precursor cells can only maintain their long-term proliferative potential and stem cell characteristics when they are cultured into neurospheres.[25],[26] The mechanism for the superior survival of spheroids is unknown. The increased cell density may enhance the survival of neurons in stringent conditions via the autocrine and/or paracrine secretion of growth factors. Lin et al. reported that the development of melanocytes in monolayers or in spheroids on a chitosan-coated surface is dependent on seeding density, and it is assumed that melanocytes at high density form spheroids via migration after close intercellular contact. They concluded that melanocyte spheroid formation needs two signals: one is chitosan as the substrate, and the other is the intercellular communication by way of contact. They also proposed that when cell–cell interactions exceed cell–substrate interactions, cells may aggregate into multicellular spheroids. In our study, cellular spheres were successfully obtained without any biomaterial as a substrate, indicating that the substrate is not a necessary condition. Melanocytes in the spheroids proliferated continuously, resulting in a gradual enlargement of the spheres, and they were capable of growing into monolayered dendritic melanocytes when they were reinoculated in culture plates.
Many recent studies have suggested that the pathways of autophagy, apoptosis and necroptosis are connected to one another as mechanisms of cell death.[27] At first, nutrient starvation, including long-term trypsinization, causes autophagy. When the starvation is prolonged, autophagy-dependent cell death occurs, and eventually apoptosis and/or necroptosis occurs. We observed some melanosome-containing autophagosomes in melanocytes derived from spheroids using transmission electron microscopy, suggesting the involvement of autophagy.
Our findings show that melanocytes have two interchangeable morphologies. Repetitive long-term trypsinizationcan induce the formation of cellular spheroids, in which the melanocytes retain the capacity of proliferation and differentiation. Melanocytes from the spheroids had a morphology similar to that of melanocyte stem cells and proliferated quickly. Hence, the formation of melanocyte spheroids by repetitive long-term trypsinization seems to be a promising method to enhance the activity and purification of melanocytes, and thus facilitate their transplantation for the treatment of vitiligo.[28] Further studies are needed to determine the clinical applications of our findings.
Acknowledgments
The authors are very grateful to Professor V.J. Hearing for help with the Englishlanguage editing.
Financial support and sponsorship
The work was supported by Foundation of the National Natural Science (No 81673078) and the Clinical Medicine Science and Technology Projects of Jiangsu (BL 2014036).
Conflicts of interest
There are no conflicts of interest.
| 1. |
Spritz RA. Modern vitiligo genetics sheds new light on an ancient disease. J Dermatol 2013;40:310-8.
[Google Scholar]
|
| 2. |
Colucci R, Dragoni F, Moretti S. Oxidative stress and immune system in vitiligo and thyroid diseases. Oxid Med Cell Longev 2015;2015:631927.
[Google Scholar]
|
| 3. |
Allam M, Riad H. Concise review of recent studies in vitiligo. Qatar Med J 2013;2013:1-9.
[Google Scholar]
|
| 4. |
Ebrahimi A, Radmanesh M, Kavoussi H. Recipient site preparation for epidermal graft in stable vitiligo by a special fraise. An Bras Dermatol 2015;90:55-60.
[Google Scholar]
|
| 5. |
Lin SJ, Jee SH, Hsiao WC, Yu HS, Tsai TF, Chen JS, et al. Enhanced cell survival of melanocyte spheroids in serum starvation condition. Biomaterials 2006;27:1462-9.
[Google Scholar]
|
| 6. |
Lin SJ, Jee SH, Hsaio WC, Lee SJ, Young TH. Formation of melanocyte spheroids on the chitosan-coated surface. Biomaterials 2005;26:1413-22.
[Google Scholar]
|
| 7. |
Shigemoto T, Kuroda Y, Wakao S, Dezawa M. A novel approach to collecting satellite cells from adult skeletal muscles on the basis of their stress tolerance. Stem Cells Transl Med 2013;2:488-98.
[Google Scholar]
|
| 8. |
Popovic A, Wiggins T, Davids LM. Differential susceptibility of primary cultured human skin cells to hypericin PDT in anin vitro model. J PhotochemPhotobiol B 2015;149:249-56.
[Google Scholar]
|
| 9. |
Godwin LS, Castle JT, Kohli JS, Goff PS, Cairney CJ, Keith WN, et al. Isolation, culture, and transfection of melanocytes. CurrProtoc Cell Biol 2014;63:1.8.1-20.
[Google Scholar]
|
| 10. |
Lennon DP, Schluchter MD, Caplan AI. The effect of extended first passage culture on the proliferation and differentiation of human marrow-derived mesenchymal stem cells. Stem Cells Transl Med 2012;1:279-88.
[Google Scholar]
|
| 11. |
Kuroda Y, Kitada M, Wakao S, Nishikawa K, Tanimura Y, Makinoshima H, et al. Unique multipotent cells in adult human mesenchymal cell populations. Proc Natl AcadSci U S A 2010;107:8639-43.
[Google Scholar]
|
| 12. |
Dilwali S, Patel PB, Roberts DS, Basinsky GM, Harris GJ, Emerick KS, et al. Primary culture of human Schwann and schwannoma cells: Improved and simplified protocol. Hear Res 2014;315:25-33.
[Google Scholar]
|
| 13. |
Li QL, Wu YH, Niu M, Lu XJ, Huang YH, He DH, et al. Protective effects of tacalcitol against oxidative damage in human epidermal melanocytes. Int J Dermatol 2017;56:232-8.
[Google Scholar]
|
| 14. |
Liu Q, Zhang RZ, Li D, Cheng S, Yang YH, Tian T, et al. Muse cells, a new type of pluripotent stem cell derived from human fibroblasts. Cell Reprogram 2016;18:67-77.
[Google Scholar]
|
| 15. |
Begemann I, Galic M. Correlative light electron microscopy: Connecting synaptic structure and function. Front Synaptic Neurosci 2016;8:28.
[Google Scholar]
|
| 16. |
Cai WX, Zheng LW, Ma L, Huang HZ, Yu RQ, Zwahlen RA, et al. Tumorigenicity and validity of fluorescence labelled mesenchymal and epithelial human oral cancer cell lines in nude mice. Biomed Res Int 2016;2016:4897986.
[Google Scholar]
|
| 17. |
Lee KS, Choe G, Nam KH, Seo AN, Yun S, Kim KJ, et al. Immunohistochemical classification of primary and secondary glioblastomas. Korean J Pathol 2013;47:541-8.
[Google Scholar]
|
| 18. |
Kormos B, Belso N, Bebes A, Szabad G, Bacsa S, Széll M, et al. In vitro dedifferentiation of melanocytes from adult epidermis. PLoS One 2011;6:e17197.
[Google Scholar]
|
| 19. |
Huang Q, Liang W, Xu D, Zhou Y, Wang T, Liang Y, et al. Ultrastructural observations of human epidermal melanocytes cultured on polyethylene terephthalate film. Micron 2013;48:49-53.
[Google Scholar]
|
| 20. |
Tang J, Li Q, Cheng B, Jing L. Primary culture of human face skin melanocytes for the study of hyperpigmentation. Cytotechnology 2014;66:891-8.
[Google Scholar]
|
| 21. |
Adema GJ, de Boer AJ, van 'tHullenaar R, Denijn M, Ruiter DJ, Vogel AM, et al. Melanocyte lineage-specific antigens recognized by monoclonal antibodies NKI-beteb, HMB-50, and HMB-45 are encoded by a single cDNA. Am J Pathol 1993;143:1579-85.
[Google Scholar]
|
| 22. |
Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nat Cell Biol 2010;12:747-57.
[Google Scholar]
|
| 23. |
Birlea SA, Costin GE, Roop DR, Norris DA. Trends in regenerative medicine: Repigmentation in vitiligo through melanocyte stem cell mobilization. Med Res Rev 2017;37:907-35.
[Google Scholar]
|
| 24. |
Shigemoto T, Kuroda Y, Wakao S, Dezawa M. A novel approach to collecting satellite cells from adult skeletal muscles on the basis of their stress tolerance. Stem Cells Transl Med 2013;2:488-98
[Google Scholar]
|
| 25. |
Li YC, Tsai LK, Wang JH, Young TH. A neural stem/precursor cell monolayer for neural tissue engineering. Biomaterials 2014;35:1192-204.
[Google Scholar]
|
| 26. |
Chen JH, Lee DC, Chen MS, Ko YC, Chiu IM. Inhibition of neurosphere formation in neural stem/progenitor cells by acrylamide. Cell Transplant 2015;24:779-96.
[Google Scholar]
|
| 27. |
Goodall ML, Cramer SD, Thorburn A. Autophagy complexes cell death by necroptosis. Oncotarget 2016;7:50818-9.
[Google Scholar]
|
| 28. |
Bae JM, Choi KH, Jung HM, Kim SY, Kim M, Kim GM, et al. Subsequent vitiligo after hematopoietic stem cell transplantation: A nationwide population-based cohort study from korea. J Am AcadDermatol 2017;76:459-63.
[Google Scholar]
|
Fulltext Views
4,239
PDF downloads
1,052





![[Figure - 1]](#fig_ijdvl_2019_85_3_258_252577_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2019_85_3_258_252577_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2019_85_3_258_252577_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2019_85_3_258_252577_f4.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2019_85_3_258_252577_f5.jpg){kind=link}