Translate this page into:
Muir-Torre syndrome
2 Department of Biotechnology, Govind Ballabh Pant Engineering College, Pauri, Uttarakhand, India
Correspondence Address:
Ashutosh Kansal
Department of Pathology, Institute of Nuclear Medicine and Allied Sciences, Defence Research and Development Organization, Brig, S. K. Majumdar Road, Timarpur, New Delhi - 110 054
India
| How to cite this article: Kansal A, Aggarwal S, Gupta D. Muir-Torre syndrome. Indian J Dermatol Venereol Leprol 2014;80:356-358 |
Sir,
Muir-Torre syndrome (MTS) is a rare autosomal dominantly inherited genodermatosis involving sebaceous neoplasms which occur as markers for multiple internal malignancies. Diagnostic criteria include presence of at least one sebaceous gland neoplasm and synchronous or metachronous occurrence of at least one internal organ cancer in the absence of other precipitating factors such as radiotherapy or acquired immunodeficiency syndrome (AIDS). [1]
A 65-year-old male patient presented with a slowly increasing, subcutaneous, nodular lesion on the back. The patient noticed the swelling in 2002, but did not seek any medical consultation as the lesion was asymptomatic. [Figure - 1]a and b The lesion was excised under local anesthesia. We received a well-circumscribed, gray-white soft tissue mass measuring 4 × 3.5 × 3.5 cm. Cut section revealed multiple small cystic areas.
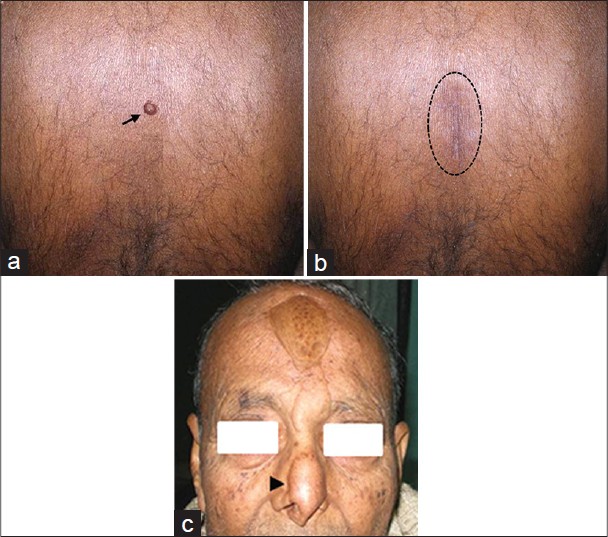 |
| Figure 1: (a) Lesion on the back (arrow), clinically diagnosed as sebaceous cyst, (b) postoperative with healed scar (dotted circle). (c) Reconstructed nose with pedicle graft from forehead after wide excision for squamous cell carcinoma of right ala of nose (arrow head) |
Microscopically, the lesion was composed of multiple, well-circumscribed lobules of various sizes. The lobules were made up predominantly of small, uniform basaloid cells with bland nuclear features admixed with mature-appearing sebaceous cells from the periphery to the center comparable to normal sebaceous glands [Figure - 2]a and b.
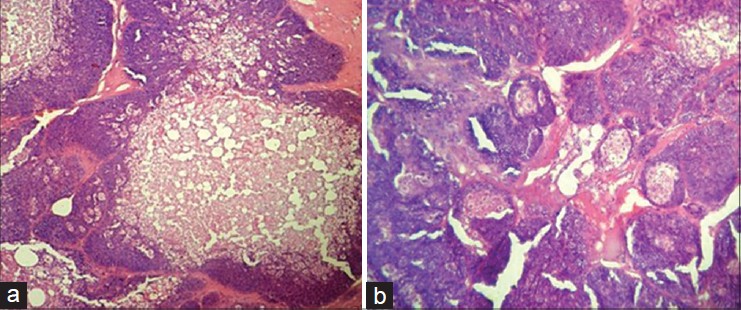 |
| Figure 2: (a and b) Subcutaneous nodule showing lobules of basaloid cells admixed with mature sebaceous cells and cystic areas (H and E, original magnification, ×10 (a) and ×40 (b)) |
The mature sebaceous cells showed abundant vacuolated cytoplasm and ovoid nuclei. The lobules contained haphazardly distributed ducts and areas of cystic degeneration filled with eosinophilic material. The lesion was reported as sebaceoma.
In view of its rarity and strong association with MTS, the patient was screened for internal malignancies. Colonoscopy revealed an ulcero-nodular mass in the cecum and ascending colon [Figure - 3]a and b.
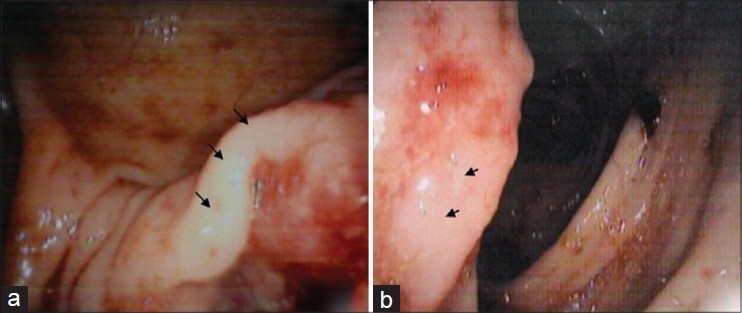 |
| Figure 3: (a and b) Colonoscopy revealing mass lesion in cecum and ascending colon (arrow) |
His hemoglobin was 10.2 gm/dl and stool was positive for occult blood by heme-spot guaiac test. Other hematological and biochemical parameters were normal. Serum carcinoembryonic antigen (CEA) level was 9.79 ng/ml (normal < 4.7 ng/ml). Computed tomography (CT) scan of the abdomen revealed irregular thickening of cecum and ascending colon suggestive of carcinoma [Figure - 4].
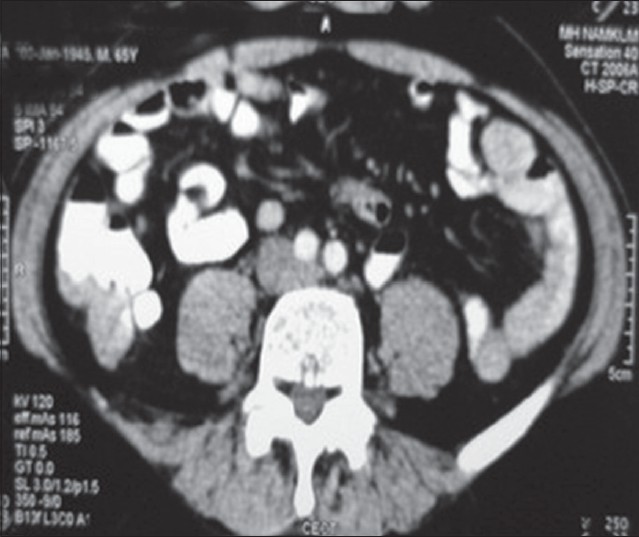 |
| Figure 4: Computed tomography scan of abdomen specially focused on cecum and ascending colon, showing irregular thickening of bowel wall |
Colonoscopic biopsy of the ascending colon mass confirmed the diagnosis of well-differentiated adenocarcimoma [Figure - 5].
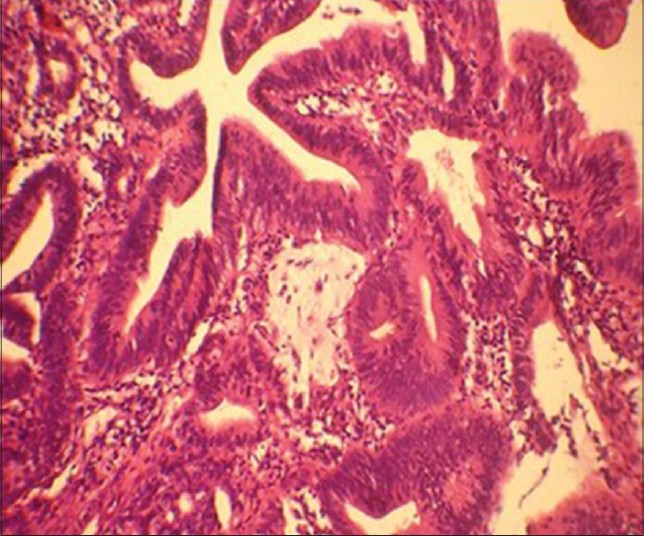 |
| Figure 5: Colonoscopic biopsy tissue showing well-differentiated adenocarcinoma ( H and E, original magnification, ×40) |
In year 2000, the patient underwent wide local excision of right ala of nose for well-differentiated squamous cell carcinoma [Figure - 1]c. There was no history of radiotherapy.
The patient has four sons and one daughter in their early forties, who are currently healthy. He has four sisters. One sister underwent a subtotal gastrectomy in her fourth decade of life for gastric cancer and another one was operated for carcinoma of the right breast at the age of 55 years. His mother had endometrial carcinoma and died in her 50s. One of his nephews died in his 30s from some cutaneous malignancy (details not available) and another one underwent right hemi-colectomy in his fourth decade for carcinoma of the ascending colon. Colonoscopy of two elder sons of the patient was performed and was within normal limits.
The MTS syndrome was independently described by Muir in 1967 and Torre in 1968 and has since been recognized as a subtype of Lynch type II hereditary non-polyposis colon cancer (HNPCC). [2] The discovery of a MTS-associated sebaceous tumor is rare and the presence of even a single lesion warrants evaluation for the syndrome. An archival review of the histopathology specimens stored in the dermatology department at the Mayo Clinic over a 60-year period found only 59 patients with one or more of these lesions. Of these 59 patients, 25 (42%) had one or more visceral malignancies. Colorectal cancer is the commonest visceral neoplasm to occur in MTS, and the most frequent initial cancer. [3] In common with other forms of HNPCC, colorectal malignancies in MTS are usually proximal in location and tend to have a more indolent course than other forms of colorectal cancer.
Akhtar et al., identified a total of 205 reported cases in the world literature. Fifty-one percent of MTS patients develop at least one colorectal cancer. Genitourinary malignancies, mainly transitional cell carcinomas, occur in 24% of individuals. Carcinomas of the endometrium, ovary, breast, parotid, upper gastrointestinal tract, larynx, and hematological malignancies are also associated with MTS. Fifty-six percent of skin lesions in MTS occur after diagnosis of the first malignancy, 6% occur concomitantly and 22% occur as the first malignancy of the syndrome. [4] Cutaneous lesions may occur as long as 25 years before or 37 years after the internal malignancy. [3] The genetic disorder in MTS is an inherited germline mutation in one of the DNA mismatch repair genes, hMSH1 and hMSH2. [5] It is inherited with a high degree of penetrance and variable expression. The syndrome occurs in both sexes, with a male to female ratio of 3:2. Children of an MTS individual have a 50% risk of inheriting the predisposition to developing cancers. Therefore, screening recommendations for patients with MTS and their first-degree relatives include yearly examination of the breast in women and testicles and prostate in men, colonoscopy every 1-2 years from age 25 years or 5 years before the youngest age of diagnosis of colorectal carcinoma in the family and annually from age 40 years. Annual pelvic examination with transvaginal ultrasonography and endometrial biopsy in women with a gene mutation, from age 25 years are recommended. Laboratory tests including complete blood count, tumor markers, for example, CA-125 and carcinoembryonic antigen (CEA), fecal occult blood, and urinalysis should be done at least once in a year for early detection of malignancy. [6]
Since the carcinomas occurring as part of MTS have an indolent course and relatively good prognosis, surgical removal of primary and metastatic cancer may be curative. [3] The combination of interferon (IFN-alpha2a) with retinoids (isotretinoin) seems to be of promise to prevent tumor development in MTS.
| 1. |
Schwartz RA, Torre DP. The Muir-Torre syndrome: A 25-year retrospect. J Am Acad Dermatol 1995;33:90-104.
[Google Scholar]
|
| 2. |
Barana D, vander Klift H, Wijnen J, Longa ED, Radice P, Cetto GL, et al. Spectrum of genetic alterations in Muir-Torre syndrome is the same as in HNPCC. Am J Med Genet A 2004;125:318-9.
[Google Scholar]
|
| 3. |
Cohen PR, Kohn SR, Kurzrock R. Association of sebaceous gland tumours and internal malignancy: The Muir-Torre syndrome. Am J Med 1991;90:606-13.
[Google Scholar]
|
| 4. |
Akhtar S, Oza KK, Khan SA, Wright J. Muir-Torre syndrome: Case report of a patient with concurrent jejunal and ureteral cancer and a review of the literature. J Am Acad Dermatol 1999;41:681-6.
[Google Scholar]
|
| 5. |
Suspiro A, Fidalgo P, Cravo M, Albuquerque C, Ramalho E, Leitão CN, et al. The Muir-Torre syndrome: A rare variant of hereditary non-polyposis colorectal cancer associated with hMSH2 mutation. Am J Gastroenterol 1998;93:1572-4.
[Google Scholar]
|
| 6. |
Cohen PR, Kohn SR, Davis DA, Kurzrock R. Muir-Torre syndrome. Dermatol Clin 1995;13:79-89.
[Google Scholar]
|
Fulltext Views
2,634
PDF downloads
1,069





![[Figure - 1]](#fig_ijdvl_2014_80_4_356_136928_u1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2014_80_4_356_136928_u2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2014_80_4_356_136928_u3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2014_80_4_356_136928_u4.jpg){kind=link}
![[Figure - 5]](#fig_ijdvl_2014_80_4_356_136928_u5.jpg){kind=link}