
Translate this page into:
Multiple subcutaneous nodules with delayed developmental milestones in a child
Corresponding author: Dr. Anupama Bains, Department of Dermatology, Venereology & Leprology, All India Institute of Medical Sciences, Basni, Jodhpur, Rajasthan, India. whiteangel2387@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Sadhukhan S, Bains A, Khera D, Aggarwal D. Multiple subcutaneous nodules with delayed developmental milestones in a child. Indian J Dermatol Venereol Leprol. 2025;91:117-9. doi: 10.25259/IJDVL_1395_2023
A 5-year-old girl born to second-degree consanguineous parents presented with multiple skin-coloured subcutaneous nodules of four years duration. The lesions started on the fingers of the right hand and subsequently progressed to involve remaining fingers and toes. The child initially attained all developmental milestones till six months of age, but now developed neuroregression syndrome. The child was unable to sit without support, stand with support or crawl and speak bi-syllables. She also had myoclonic jerks and developmental dysplasia of the right hip (DDH). There was a history of similar complaints in one elder cousin who died at the age of six. In antenatal history, the mother had obstructed labour and the child needed resuscitation at birth because of meconium-stained liquor and also had caput succedaneum.
On examination, multiple discrete firm subcutaneous skin-coloured nodules ranging from 0.3 to 1 cm in diameter were present on hands and feet, predominantly on proximal and distal interphalangeal joints (PIP and DIP) with decreased mobility of joints and fixed flexion deformity of interphalangeal joints [Figure 1]. Multiple punctate corneal epithelial-stromal dystrophies were noticed on both eyes through slit lamp examination. A meningocele of 3 × 3 cm in diameter was observed with overlying erythema on the sacrococcygeal region in the midline, with positive transillumination and surrounded by a few firm small nodules. Complete haemogram and serum biochemical parameters were normal. Magnetic resonance imaging of the brain showed mild cerebral atrophy, with prominence of the ventricular system and sulcal spaces. Radiography showed deformities of the interphalangeal joints with soft tissue swelling. A biopsy was obtained from a nodule on the left little toe in which the deeper dermis showed sclerotic nodule containing distended foamy histiocytes and sparse lymphocytes. Periodic acid-schiff (PAS)-alcian blue stain showed alcian blue-positive intracytoplasmic material and PAS-positive deposits in macrophages. A mild perivascular lymphocytic infiltrate was seen in the upper dermis. No pathological changes were seen in the overlying epidermis covered with thick compact orthokeratotic stratum corneum, appropriate for the anatomic site [Figures 2a–2c].
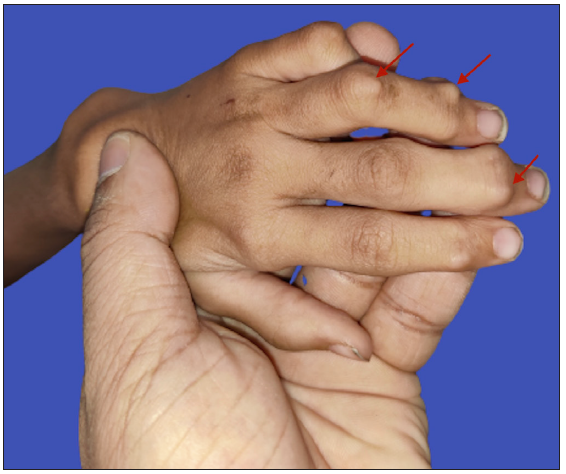
- Skin coloured firm non-tender nodules (red arrows) on proximal and distal interphalangeal joints of the left hand with flexion deformity.
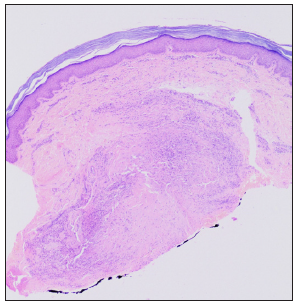
- Sclerotic nodule in the deep dermis and overlying normal epidermis (Haematoxylin and eosin, 20x).
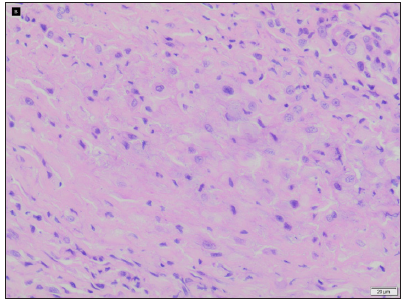
- Foamy histiocytes and sparse lymphocytes in sclerotic nodules (Haematoxylin and eosin, 400x).
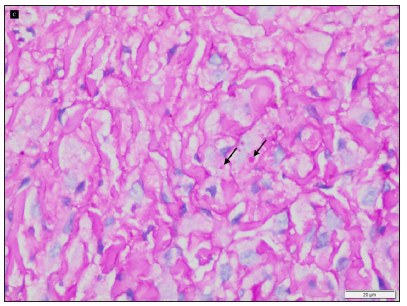
- Intracytoplasmic alcian blue positive material and periodic acid schiff positive deposits (black arrows) (periodic acid schiff-alcian blue stain with diastase, 400x).
Question
What is your diagnosis?
Answer
Diagnosis
Farber disease
Other investigations
Whole exome sequencing (genetic analysis) revealed a missense mutation in the ASAH1 gene. The variation was the amino acid substitution of arginine for tryptophan at codon 185 in exon 8 (c.553T>C; p.Trp185Arg) and a diagnosis of Farber disease (FD) was made.
Discussion
FD is a lysosomal storage disorder caused by acid ceramidase (EC3.5.1.23) deficiency, which deacetylates ceramide to sphingosine and free fatty acid, and is deficient as a result of these mutations and lead to widespread ceramide accumulation, affecting various organs and accompanying clinical symptoms.1 FD is divided into seven subgroups, including the classical variant type 1, intermediate type 2, mild type 3, neonatal-visceral variant type 4, neurological progressive variant type 5, combined FD and Sandhoff disease type 6 and prosaposin deficiency type 7.1 Subcutaneous nodules, arthritis and hoarseness of voice are the typical triad of symptoms that appear in FD.2 Among the ocular findings, the cherry red spot in the macula is the most typical finding and others are retinal and corneal opacities and macular degeneration.3 Pathologically, the subcutaneous nodules consist of granulomatous lesions that show an array of histological changes, depending on the stage of the disease. The majority of the cells are spindle-shaped at first, but as the stage advances on, these cells become larger and more rounded, become foam cells and collagen fibres start to proliferate and form granulomatous lesions, which are characterised by dense collagen fibres surrounding typical foam cells.4 Amirhakimi et al. discovered lymphocytes and plasma cells in areas of early fibrosis.5 In the most advanced stage, these collagen fibres can be hyalinised. The results of the histochemical stains have varied, depending on whether frozen or paraffin sections were utilised. The early-stage intracytoplasmic PAS-positive material is resistant to extraction with hot pyridine, hyaluronidase and diastase digestion. These materials, which are positive for colloidal iron and alcian blue staining, are assumed to represent non-sulfated acid mucopolysachharides and glycopeptides. A weak toluidine blue reaction (pH 4.1) demonstrates metachromasia. These materials could also be positive with Baker’s reaction for phospholipid, sudan IV, sudan black B and oil red O, thereby suggesting that thay may be ceramides.4,5 Semi-curvilinear inclusions, commonly referred to as Farber bodies, zebra bodies or banana bodies can be seen using electron microscopy in lung histiocytes, hepatocytes, endothelial cells and Kupffer cells.6 These bodies are curvilinear bodies within the cytoplasm of fibroblasts and occasionally of endothelial cells. They are also found within phagosomes of histiocytes at various stages of degradation.7
In our case, the girl exhibited classic symptoms like subcutaneous nodules, arthritis along with corneal dystrophy, features of neuroregression, myoclonic jerks and DDH. She also experienced stridor but without associated hoarseness in her voice, and therefore, the typical triad was not present. DDH is described commonly with cerebral palsy patients but it has not described with FD previously in the literature.8 Cherry red spots in the macula were absent in this case. Other disorders like stiff skin syndrome, juvenile hyaline fibromatosis and multicentric reticulohistiocytosis that may also present with subcutaneous nodules and joint contractures must be excluded.
There is currently no specific enzyme replacement therapy for FD; instead, symptomatic and supportive treatment are the mainstays of care. Early diagnosis and treatment are crucial. Before the beginning of neurological symptoms, haematopoietic stem cell transplantation may be beneficial.9
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of AI-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Acid ceramidase deficiency: Farber disease and SMA-PME. Orphanet J Rare Dis. 2018;13:121.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Acid ceramidase deficiency: Farber lipogranulomatosis and spinal muscular atrophy associated with progressive myoclonic epilepsy. In: Rosenberg RNPJ, ed. Rosenberg’s molecular and genetic basis of neurological and psychiatric disease. London: Elsevier; 2014. p. :395-402.
- [Google Scholar]
- Retinopathy in a case of Farber’s lipogranulomatosis. Arch Ophthalmol. 1966;75:752-7.
- [CrossRef] [PubMed] [Google Scholar]
- Farber’s disease (disseminated lipogranulomatosis)—a pathological, histochemical and ultrastructural study. Acta Pathol Jpn. 1979;29:135-55.
- [CrossRef] [PubMed] [Google Scholar]
- Familial lipogranulomatosis (Farber’s disease) Clin Genet. 1976;9:625-30.
- [CrossRef] [PubMed] [Google Scholar]
- Farber’s disease: A fine structural study. Ultrastruct Pathol. 1987;11:397-403.
- [CrossRef] [PubMed] [Google Scholar]
- Named bodies in dermatology. Indian J Dermatol Venereol Leprol. 2010;76:206-12.
- [CrossRef] [PubMed] [Google Scholar]
- A classification system for hip disease in cerebral palsy. Deve Med Child Neurol. 2009;51:183-92.
- [CrossRef] [Google Scholar]
- Farber disease: Clinical presentation, pathogenesis and a new approach to treatment. Pediatr Rheumatol Online J. 2007;5:15.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




