
Translate this page into:
Neonatal ichthyosis and sclerosing cholangitis (NISCH) syndrome with a novel Claudin-1 (CLDN1) mutation: A report from India
Corresponding author: Dr. Jerene Mathews, Department of Dermatology, Believers Church Medical College and Hospital, Kuttapuzha, Thiruvalla, India. jerenemathews7@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Mathews J, Chandrasekaren A. Neonatal ichthyosis and sclerosing cholangitis (NISCH) syndrome with a novel Claudin-1 (CLDN1) mutation: A case report from India. Indian J Dermatol Venereol Leprol. 2025;91:89-91. doi 10.25259/IJDVL_186_2023.
Dear Editor,
Mutations in the Claudin-1 (CLDN1, Gene ID: 9076) gene are known to result in the autosomal-recessive neonatal ichthyosis-sclerosing cholangitis (NISCH) syndrome (OMIM: 607626).1 The long-term prognosis depends on the severity of the liver disease, which is partly dependent on the mutation present.2 Other ichthyoses with neonatal cholestasis include arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome, type 2 Gaucher’s disease, and MEDNIK syndrome (mental retardation, enteropathy, deafness, neuropathy, ichthyosis and keratodermia).3
An 8-year-old boy, born of a non-consanguineous marriage, in Kerala (India), presented with dry, scaly skin, itching and poor growth of scalp hair since birth. He had cholestasis in the neonatal period, which resolved spontaneously in infancy. There was no history of similar illnesses or infant deaths in the family.
On examination, generalised ichthyosis was noted with medium-sized, dark-brown, adherent scales [Figures 1 and 2]. The central aspect of the face, cubital fossae, popliteal fossae, axillae, palms, soles and nails were spared. The pinnae, neck and pre-auricular area were affected. The scalp hair was short and wiry. Eyebrows, eyelashes and body hair were sparse. Dental enamel was hypoplastic [Figure 3]. The face was elongated with a pear-shaped nose, a bulbous nose-tip and large ears. There was no erythema, ectropion or eclabium. There was no delay in growth, hearing defects, ocular defects or cryptorchidism.
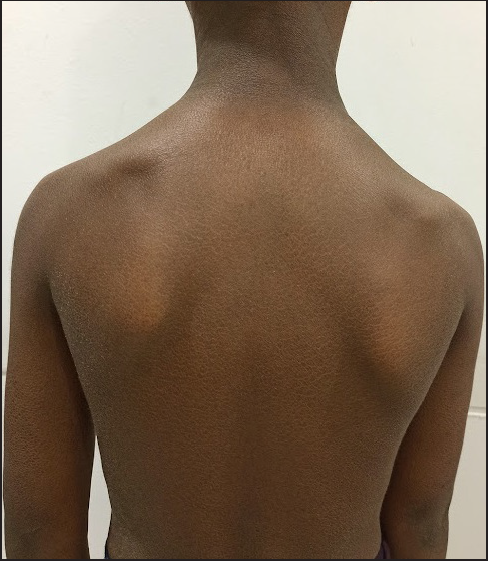
- Diffuse scaling over the trunk and upper limbs. Large ears were also present.
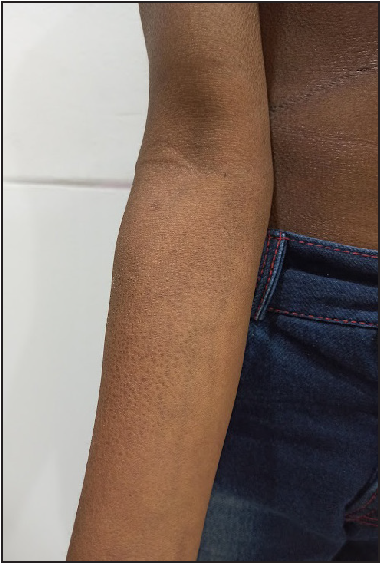
- Dark-brown, adherent scales over the forearm with relative sparing of the cubital fossa.
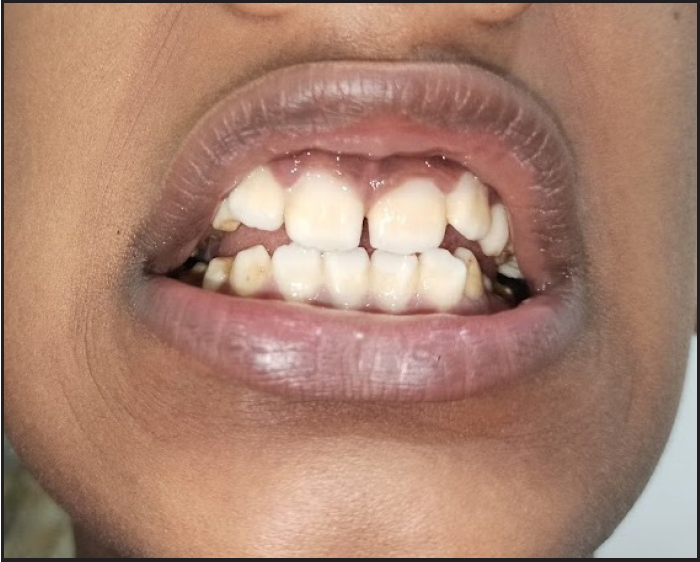
- Hypoplastic dental enamel.
Ultrasound examination of the abdomen showed course echotexture of the liver. Liver size, biliary radicals, the common bile duct and portal veins were normal. No leucocyte vacuoles were present in a peripheral smear examination. A skin biopsy revealed acanthosis, hyperkeratosis and mild hypogranulosis, but no keratinocyte vacuoles. Liver function tests, TSH and blood sugars were normal. Informed written consent was obtained from the father for genetic testing. An exome-sequencing (MedGenome Labs Ltd., Bangalore) revealed a homozygous single-base-pair deletion in Exon 1 of the CLDN1 gene (NM_021101.5:c.141del), causing a frameshift and premature truncation of the claudin-1 protein downstream to codon 47 (p.Tyr47Ter). The variant was detected at a sequencing depth of 122x and deletion of a guanidine nucleotide was seen in all reads at the chromosomal coordinate chr3:190322066 [Figure 4]. In silico analysis done using Mutation Taster reported the mutation as damaging. In addition, premature protein truncation and loss of function are known disease mechanisms in NISCH syndrome.2,4,5 This variant is not reported in gnomAD and gnomAD databases. This is a novel nucleotide change. A different nucleotide change, resulting in a similar nonsense change at the protein level (Y47X) has been documented in the ClinVar database [VCV001322095.4]. Sanger validation of the mutation in the patient and genetic testing of the parents were not done because of financial constraints.
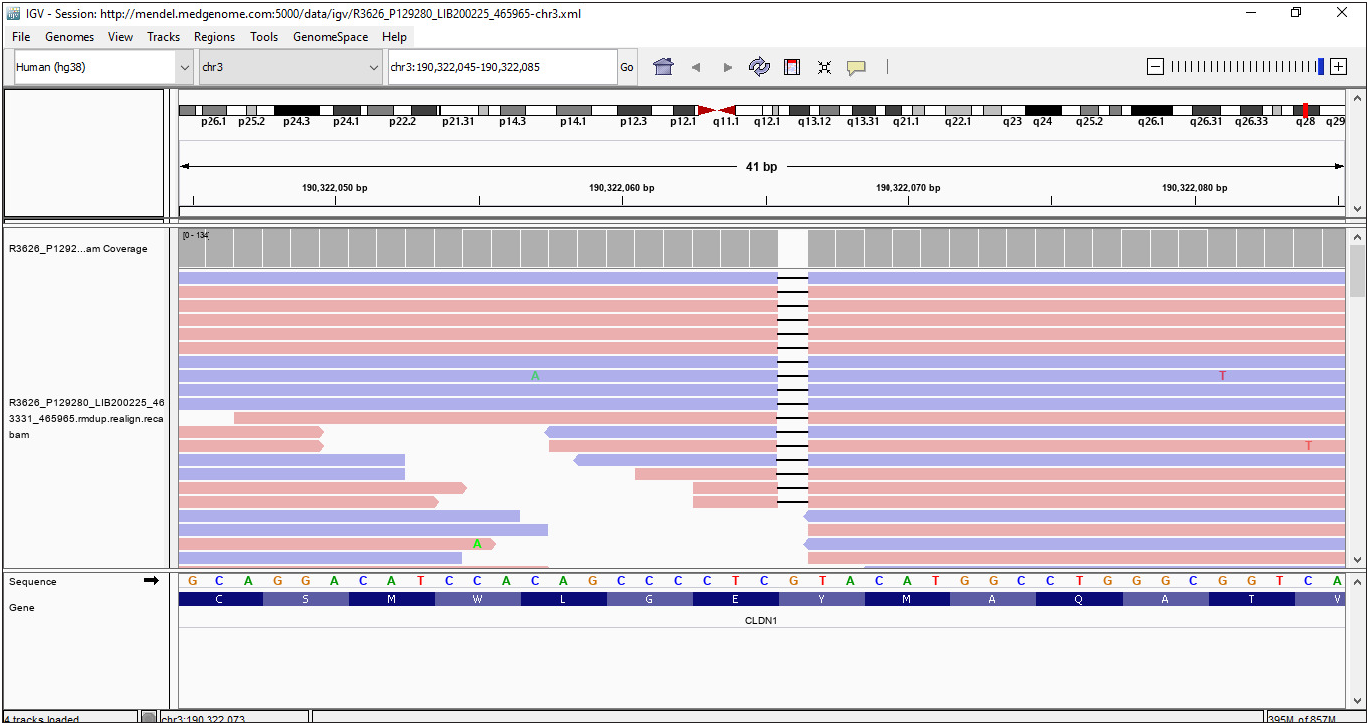
- Integrated genome viewer image for the base-pair deletion in Exon 1 of the CLDN1 gene (c.141del).
Claudin-1 is a component of tight junctions in both the epidermis and bile ducts. Defective tight junctions result in the loss of the barrier function of the epidermis. In the liver, leakage of bile from the biliary canaliculi leads to inflammation, fibrosis and obliteration of the bile ducts.2
Cases of NISCH syndrome with confirmed CLDN1 mutations have been reported in Morocco (c.200_201delTT), Switzerland (c.358delG), Turkey (c.181C>T, p.Gln61X), Iran (c.578C>A, p.Tyr159Ter) and India (p.Tyr47Ter). All of these mutations result in a truncated protein.2,4
Severe liver disease has not been reported with “Swiss” or “Turkish” mutations. In individuals with the “Moroccan” mutation, extremely variable outcomes were described, even within the same family. Some individuals had persistent cholestasis and portal hypertension with/without liver failure, eventually needing a liver transplant in the first/second decade. In others, the cholestasis was transient and resolved, leaving normal liver function.2,5 In the single case reported from India with a confirmed mutation, cholestasis resolved in infancy.4
This is the second case of NISCH with a confirmed CLDN1 mutation from India.4 The mutation detected in our patient has not been reported previously. We report this case to highlight the novel mutation and its clinical outcome.
In view of the variable severity of the hepatic disease, genotype-phenotype correlations are important to provide accurate prognostication for affected families. Because of financial constraints in genetic testing, cases of NISCH in India may be attributed to non-syndromic ichthyoses, especially when cholestasis is transient. However, it is possible that other family members may have ichthyosis with significant liver disease, as phenotypic variation has been reported within families.
Pruritus has been reported infrequently in NISCH and has been attributed to cholestasis.5 However, our patient had significant pruritus at the age of 8 years without any residual liver disease. The pruritus is probably due to the epidermal barrier defect itself, resulting from defective tight junctions.
Declaration of patient consent
Consent has been obtained from the father for publication, including the attached images.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of Artificial Intelligence (AI)-Assisted Technology for manuscript preparation
The authors confirm that there was no use of Artificial Intelligence (AI)-Assisted Technology for assisting in the writing or editing of the manuscript and no images were manipulated using the AI.
References
- Confirmation of the origin of NISCH syndrome. Hum Mutat. 2006;27:408-10.
- [CrossRef] [PubMed] [Google Scholar]
- NISCH syndrome: An extremely rare cause of neonatal cholestasis. J Hepatol. 2020;73:1257-8.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnostic approach to neonatal and infantile cholestasis: A position paper by the SIGENP liver disease working group. Dig Liver Dis. 2022;54:40-53.
- [CrossRef] [PubMed] [Google Scholar]
- NISCH Syndrome as a cause of Neonatal Cholestasis - a case report from India. J Clin Exp Hepatol. 2022;12:S79.
- [Google Scholar]
- Claudin-1 gene mutations in neonatal sclerosing cholangitis associated with ichthyosis: A tight junction disease. Gastroenterology. 2004;127:1386-90.
- [CrossRef] [PubMed] [Google Scholar]




