Translate this page into:
Neurocutaneous spectrum of multiple endocrine neoplasia-1
2 Department of Pathology, Sri Sathya Sai Institute of Higher Medical Sciences, EPIP Area, Whitefield, Bangalore-560 066, Karnataka, India
3 Department of Neurosurgery, Sri Sathya Sai Institute of Higher Medical Sciences, EPIP Area, Whitefield, Bangalore-560 066, Karnataka, India
4 Department of Radiodiagnosis, Sri Sathya Sai Institute of Higher Medical Sciences, EPIP Area, Whitefield, Bangalore-560 066, Karnataka, India
Correspondence Address:
Shireen Furtado
Consultant Dermatologist, Sri Sathya Sai Institute of Higher Medical Sciences, EPIP Area, Whitefield, Bangalore - 560 066
India
| How to cite this article: Furtado S, Ghosal N, Furtado SV, Gupta K, Hegde AS. Neurocutaneous spectrum of multiple endocrine neoplasia-1. Indian J Dermatol Venereol Leprol 2012;78:93-96 |
Abstract
Multiple endocrine neoplasia type I or Wermer syndrome is characterized by primary hyperparathyroidism, enteropancreatic endocrine tumor, and a pituitary pathology. A 35-year-old male presented with visual field defects, hyperprolactinemia, and hypogonadism. He also had multiple infraumbilical skin-colored nodules. A syndromal association of Wermer syndrome was derived using the dermal, pituitary, parathyroid, and gastrointestinal hormonal manifestations of the tumor. The radiological and histological findings of lesion which underwent biopsy are discussed. The presence of collagenomas, lipomas, and hypopigmented macules in a patient with neuroendocrine symptoms should raise the suspicion of an underlying multiple endocrine neoplasia.Introduction
Multiple endocrine neoplasia type 1 (MEN1) has an estimated prevalence of 0.02 to 0.2 per 1 000, and is caused by mutations in the MEN1 gene on chromosome 11q13. [1],[2] The autosomal dominant inheritance pattern was first described by Wermer in 1954. [3] Wermer syndrome is infrequently reported from the Australasian region. The clinical presentation is related to dysfunctions of the pituitary, pancreas, and the parathyroid in the setting of various cutaneous markers. [4] This article highlights some cutaneous manifestations which could help entertain a high index of suspicion of MEN1, especially when the neurovisceral syndrome has not yet manifested in its clinical entirety.
Case Report
A 35-year-old male presented with decreased libido and erectile dysfunction with progressive diminution of vision over a 1-year period. On examination, he had bitemporal field cuts with a visual acuity of 20/30 in the right eye and 20/40 in the left eye. Fundus examination revealed primary optic atrophy in both eyes. The rest of his neurological examination was unremarkable. General examination revealed gynecomastia and multiple skin-colored discrete papules and infraumbilical non-tender firm, non-cystic, and depressible nodules varying in size from 0.5 to 2 cm in diameter [Figure - 1]. The skin with hair overlying the nodules was unremarkable. No other foci of nodules were seen. He did not have any other cutaneous lesions. MRI of the brain showed a sellar-suprasellar lesion causing compression of the optic chiasm [Figure - 2]. Initial serum hormone evaluation revealed hyperprolactinemia, with a value of 960 ng/ml (normal, 3-26 ng/ml). Punch biopsy of the skin nodules showed features of a collagenoma [Figure - 3]a. He underwent a trans-nasal trans-sphenoidal tumor decompression of the pituitary lesion. Histological examination of the tumor revealed a pituitary adenoma neuronal choristoma (PANCH) consisting of pituitary-adenoma cells as one component, and elongated cells with cytoplasmic extensions and neuronal cells as the other. Strong immunopositivity for prolactin in pituitary adenoma cells and S-100 in the neural choristoma component was noted [Figure - 3]b. An upper gastrointestinal endoscopy revealed features of chronic duodenal ulcers. His serum calcium level was 10.8 mg/dl (normal, 8.6-10 mg/dl). Serum parathormone level was raised; 242.2 pg/ml (normal, 14-72 pg/ml) and serum testosterone level was low; 57.4 ng/dl (normal, 286-1511 ng/dl). A contrast CT scan of the neck showed homogenously enhancing enlarged bilateral superior and inferior parathyroid glands. CT of the abdomen revealed a homogenously enhancing small lesion in the tail of the pancreas [Figure - 4] which was clinically considered to be a gastrinoma, in view of recalcitrant acid-peptic disease. This was treated medically with proton pump inhibitors. He was not operated for hypercalcemia as he was asymptomatic for the same. Post-surgery, his prolactin levels normalized to 3.00 ng/ml (2.64 - 13.13 ng/ml) and there was subjective improvement in vision. He was referred to an endocrinologist for evaluation of the pancreatic lesion.
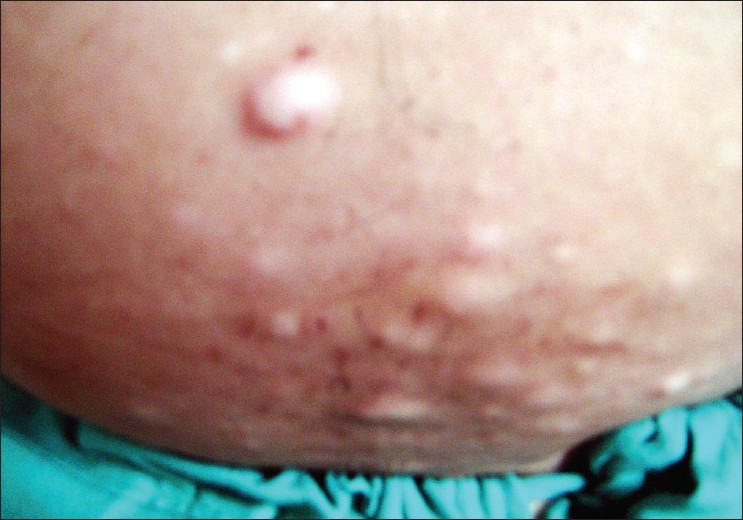 |
| Figure 1: Multiple infraumbilical skin-colored nodules representing collagenomas |
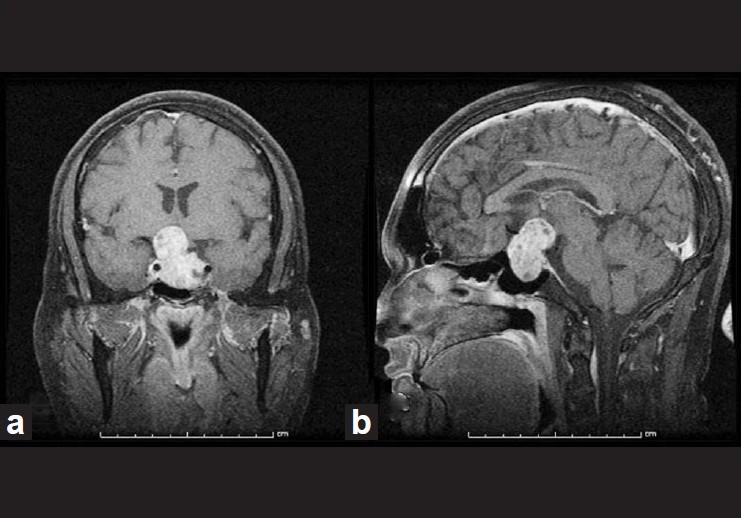 |
| Figure 2: Contrast MR images (a) coronal (b) sagittal, showing a pituitary tumor with suprasellar extension and mass effect on the optic apparatus |
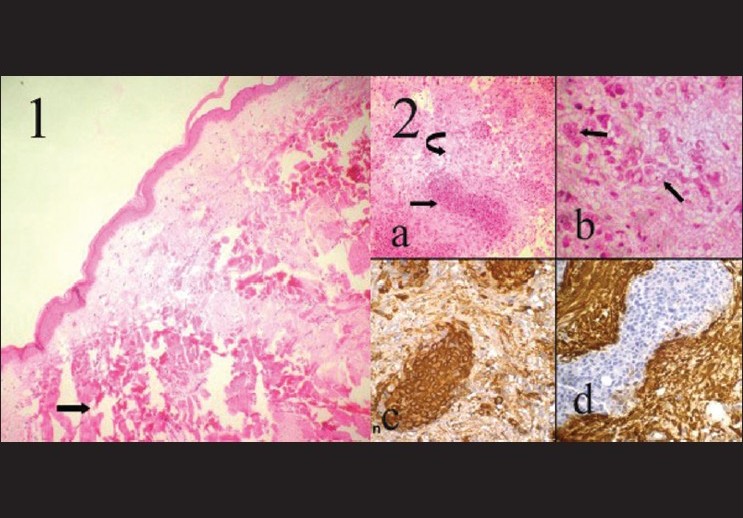 |
| Figure 3: Paraffin section of (1) collagenoma showing dense dermal collagen (arrow) [H and E, ×100] (2) Pituitary adenoma neuronal choristoma showing (a) pituitary adenoma cells (straight arrow) and elongated cells with cytoplasmic extensions with neuronal cells (curved arrow). (b) High-power view of neuronal component showing oval to polygonal cells with binucleation and prominent single nucleoli (straight arrow). (c) Immunopositivity for prolactin in pituitary adenoma cells and (d) S-100 protein positivity in the other component [H and E: (a) ×100; (b) ×400] [Avidin Biotin Complex Immunoperoxidase: (c) (d) ×400] |
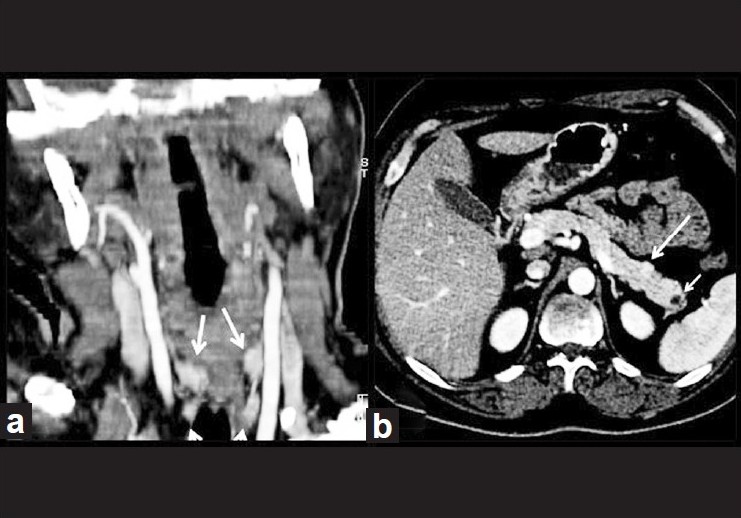 |
| Figure 4: (a) Coronal reformatted post-contrast CT images showing homogenously enhancing enlarged bilateral superior and inferior parathyroid glands (arrows). (b) Post-contrast axial abdominal CT scan showing homogenously enhancing small lesion in the tail of the pancreas (big arrow) and non-enhancing cystic lesion in the tail of pancreas (small arrow) |
Discussion
The classical P-triad (Parathyroid, Pancreatic, and Pituitary adenomas) was first reported in 1920. Wermer first suggested that the triad could represent a trait inherited in an autosomal dominant manner with high penetrance. This was based on the syndromal manifestations in a family in which a father and 4 of 9 children were affected. [3],[5]
Pituitary tumors are seen in 10 to 60% of MEN-1 patients. [5] In the absence of obvious gastrointestinal symptomatology in our patient, the association of collagenomas and the functioning pituitary tumor may well have been labeled coincidental, making the diagnosis of MEN-1 elusive. This underlines the benefit of invoking a syndromal diagnosis even in cases with partial manifestations of the syndrome. Incidentally, the association of PANCH and MEN-1 as noted in our case has anecdotal reportage, with the prolactinoma (60%) being the commonest pituitary pathology reported. [6] The other tumors are growth hormone-secreting tumors (5%), and rarely, ACTH and TSH-secreting tumors (2%). [5],[7] Parathyroid disorders are multiglandular and are seen in up to 100% of patients at 50 years of age. The gastropancreatic tract tumors include gastrinoma (40%), insulinoma (10%), VIPoma and glucagonoma (2%), foregut carcinoids (10%), and non-functioning adrenocortical tumors (20-40%). [5],[7] Symptomatic hypercalcemia due to parathyroid neoplasm is treated with subtotal or total parathyroidectomy. The management of pancreatoduodenal neuroendocrine neoplasms is controversial. An aggressive surgical approach is intended to control the functional syndromes and malignant potential for nodal or distant metastasis. [8]
MEN-1 is usually diagnosed in the 4 th decade based on endocrine symptomatology. [5] Patients with MEN-1 can present with multiple cutaneous lesions of which multiple facial angiofibromas have been reported in 88%, collagenomas in 72%, cafe΄ au lait macules in 38%, lipomas in 34%, confetti-like hypopigmented macules in 6%, and multiple gingival papules in 6%. [1],[4],[7],[9] Facial angiofibromas, also seen in tuberous sclerosis, tend to be smaller and fewer in MEN-1. They are often observed on the upper lip and the vermilion border of the lip, areas that tend to be spared in tuberous sclerosis. [1],[4] Tuberous sclerosis has associated ash-leaf macules, forehead plaques, areas of leathery skin (shagreen patches), and periungual fibromas, also known as Koenen′s tumors, besides cortical tubers in the brain, glial tumors, and tumors in the kidney, lung, and heart. [4] Neurofibromatosis-1, a phakomatosis can be differentiated from MEN-1 by the presence of cutaneous neurofibromas or a single plexiform neurofibromas on the face, cafι-au-lait macules, bone deformities like sphenoid wing dysplasia, Lisch hamartomatous nodules on the iris, and optic nerve gliomas or nerve sheath tumors like schwannomas or meningiomas. Confluent and reticulated papillomatosis, well-differentiated squamous-cell carcinoma, and superficial spreading malignant melanoma have also been reported in a patient with Wermer syndrome. [10] These cutaneous findings may be helpful in diagnosing patients with MEN-1 syndrome before the onset of endocrine manifestations of the hormone-secreting tumors. [5] The combination of multiple angiofibromas (more than three) and any collagenoma had a relative high sensitivity and excellent specificity and was identified as the best dermatological criterion to identify MEN-1 patients with cutaneous manifestations. [1] In patients with cutaneous stigmata of MEN-1, annual estimation of serum calcium, prolactin, insulin, gastrin, and blood glucose has been performed after 10 years age so that an early diagnosis of the endocrinal manifestation of the syndrome can be established. Our patient presented with gynecomastia and multiple collagenomas on the abdomen without any facial angiofibromas. This report highlights the importance of including the diverse neurocutaneous manifestations in the dermatologist′s symptom-specific diagnostic armamentarium to enable a diagnosis of the MEN-1 syndrome early-on in its clinical course.
| 1. |
Asgharian B, Turner ML, Gibril F, Entsuah LK, Serrano J, Jensen RT. Cutaneous tumors in patients with multiple endocrine neoplasm type 1 (MEN1) and gastrinomas: Prospective study of frequency and development of criteria with high sensitivity and specificity for MEN1. J Clin Endocrinol Metab 2004;89:5328-36.
[Google Scholar]
|
| 2. |
O'Brien T, O'Riordan DS, Gharib H, Scheithauer BW, Ebersold MJ, van Heerden JA. Results of treatment of pituitary disease in multiple endocrine neoplasia, type I. Neurosurgery 1996;39:273-8.
[Google Scholar]
|
| 3. |
Glascock MJ, Carty SE. Multiple endocrine neoplasia type 1: Fresh perspective on clinical features and penetrance. Surg Oncol 2002;11:143-50.
[Google Scholar]
|
| 4. |
Darling TN, Skarulis MC, Steinberg SM, Marx SJ, Spiegel AM, Turner M. Multiple facial angiofibromas and collagenomas in patients with multiple endocrine neoplasia type 1. Arch Dermatol 1997;133:853-7
[Google Scholar]
|
| 5. |
Falchetti A, Marini F, Luzi E, Giusti F, Cavalli L, Cavalli T, et al. Multiple endocrine neoplasia type 1 (MEN1): Not only inherited endocrine tumors. Genet Med 2009;11:825-35
[Google Scholar]
|
| 6. |
Walia R, Bhansali A, Kumar A, Behera A, Bhadada S, Dutta P. Multiple endocrine neoplasia type-1: An unusual presentation. Endocrinologist 2009;19:164-6.
[Google Scholar]
|
| 7. |
Trouillas J, Labat-Moleur F, Sturm N, Kujas M, Heymann MF, Figarella-Branger D, et al. Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): A case-control study in a series of 77 patients versus 2509 non-MEN1 patients. Am J Surg Pathol 2008;32:534-43.
[Google Scholar]
|
| 8. |
Hausman MS Jr, Thompson NW, Gauger PG, Doherty GM. The surgical management of MEN-1 pancreatoduodenal neuroendocrine disease. Surgery 2004;136:1205-11.
[Google Scholar]
|
| 9. |
Hoang-Xuan T, Steger JW. Adult-onset angiofibroma and multiple endocrine neoplasia type I. J Am Acad Dermatol 1999;41:890-2.
[Google Scholar]
|
| 10. |
Baldauf C, Vortmeyer AO, Koch CA, Sticherling M. Combination of multiple skin malignancies with multiple endocrine neoplasia type 1: Coincidental or pathogenetically related? Dermatology 2009;219:365-7.
[Google Scholar]
|
Fulltext Views
3,478
PDF downloads
1,372





![[Figure - 1]](#fig_ijdvl_2012_78_1_93_90956_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2012_78_1_93_90956_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2012_78_1_93_90956_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2012_78_1_93_90956_f4.jpg){kind=link}