Translate this page into:
Nevoid basal cell carcinoma syndrome (Gorlin-Goltz syndrome)
Correspondence Address:
Senda Majdoub
Department of Radiology, CHUFarhat Hached, Sousse, 4000
Tunisia
| How to cite this article: Majdoub S, Zaghouani H, Cheikh YB, Laadhari M, Amara H, Abassi D, Kraeim C. Nevoid basal cell carcinoma syndrome (Gorlin-Goltz syndrome). Indian J Dermatol Venereol Leprol 2015;81:414-416 |
Sir,
Nevoid basal cell carcinoma syndrome (NBCCS), also known as Gorlin-Goltz syndrome, is an infrequent multisystemic disease with a spectrum of developmental anomalies and predisposition to a range of neoplasms. [1] Other clinical manifestations frequently seen are hyperkeratosis of jaw, pits on the palms and toes, bifid ribs, skeletal abnormalities, intracranial ectopic calcifications and facial dysmorphism. [1],[2]
A 45-year-old man presented with multiple asymptomatic pigmented lesions over his face and chest, all of which had been present for 10 years. These lesions were initially small and gradually progressed. The patient had also noticed an increase in the number in the recent past. There was no history of bleeding from the lesions, seizures, loss of appetite or loss of weight. None of the family members had similar complaints. On examination, multiple discrete, darkly pigmented, smooth- to rough-surfaced papules and plaques of varying sizes were noted over the face [Figure - 1]. Numerous tiny pits were evident on his palms and soles. Hair shaft examination was normal. Intraoral examination showed jaw hyperkeratosis and revealed missing right and left mandibular first and second molars.
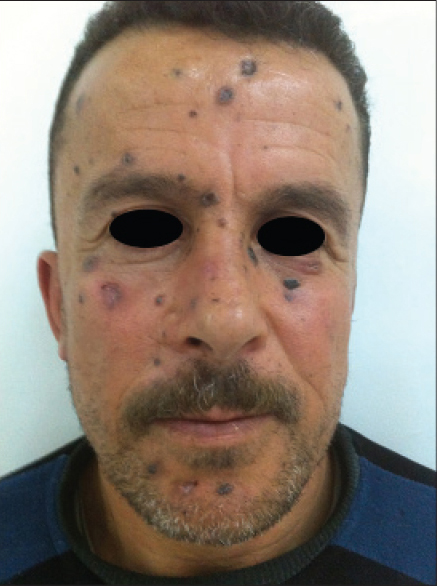 |
| Figure 1: Multiple basal cell carcinomas on the face |
With these clinical findings a probable diagnosis of Gorlin syndrome or basal cell nevus syndrome was made and further investigations were carried out.
A panoramic radiograph revealed well-defined, multiple, unilocular radiolucencies with sclerotic borders in the mandibular body, ramus and symphysis areas. Computed tomography (CT) images showed hypodense areas in relation to the left mandibular body separated by hyperdense septae. A minor breach in cortical integrity was observed in the lingual aspect of the ramus region. A single lytic lesion was observed in the symphysis region. The lesion involved the right half of the maxilla including the maxillary sinus [Figure - 2]. Calcification of falx cerebri and tentorium cerebella was also observed [Figure - 3].
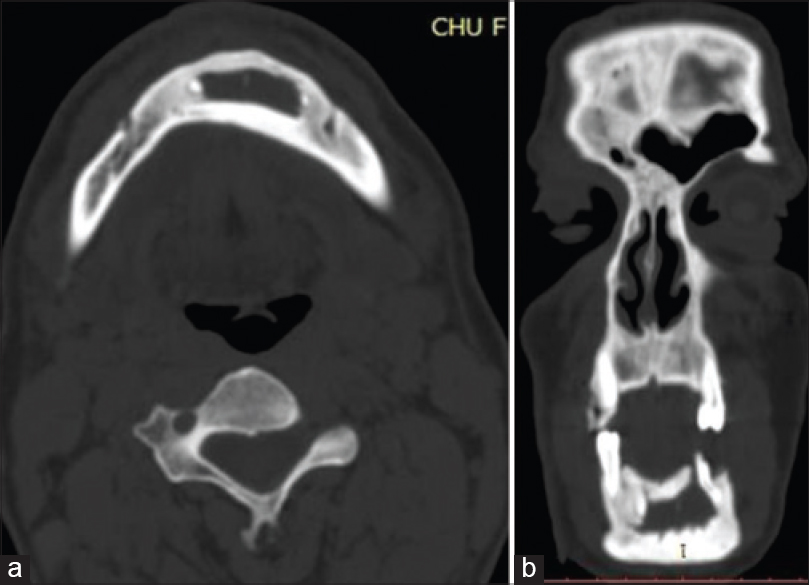 |
| Figure 2: (a) Well-defined multiple unilocular hypodense areas with sclerotic borders in the mandibular body, (b) ramus and symphysis areas. The largest one was located in the body of the mandible on the left side |
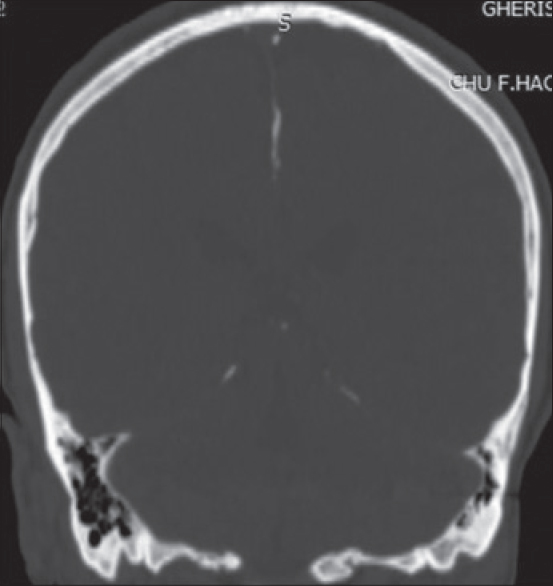 |
| Figure 3: Calcifications of falx cerebri and tentorium cerebella |
These investigative findings were consistent with our clinical diagnosis of Gorlin syndrome.
Histopathological examination of a representative specimen from one of the pigmented papules showed tumor nests consistent with basal cell carcinoma.
Nevoid basal cell carcinoma syndrome is a rare, complex genetic disorder characterised by a wide variety of developmental abnormalities and a predisposition to developing certain forms of cancer, particularly a type of skin cancer known as basal cell carcinoma. The diagnostic criteria for this syndrome were first established by Evans et al. [3] and later modified by Kimonis et al. in 1997. [4] The syndrome is established when at least two major or one major and two minor criteria are present [Table - 1].

The condition is inherited as an autosomal dominant trait with a high level of penetrance and variable expressiveness that can manifest itself spontaneously. [1] Mutations in the tumour suppressor gene called patched (PTCH1), located in the 9q22.3-q31 chromosome, have been identified as the cause of this syndrome. [1]
Multiple odontogenic keratocysts (kcots) are the most consistent and representative sign of nevoid basal cell carcinoma syndrome in the first and second decades of life. [2] Multiple keratocysts may be present in other conditions, such as orofacial digital syndrome, Ehlers−Danlos syndrome, Noonan syndrome and Simpson-Golabi-Behmel syndrome. [5] The keratocysts seen in this syndrome are multiple, ranging in number from 1 to 30, occur in the first decade of life and have a higher rate of recurrence. Clinically, the most common site is the mandibular molar ramus region and the condition is usually asymptomatic until the keratocysts reach a large size when they present with swelling, mild pain and displaced or impacted teeth.
Radiographic features include radiolucent lesions, unilocular or multilocular, with smooth/scalloped borders, associated with impacted/displaced teeth and a tendency to grow along the internal aspect of the jaws causing minimal expansion.
On computed tomography examination, keratocysts appear as minimally expansile benign lesions with scalloped borders and high attenuation of contents which do not enhance with contrast.
Keratocysts in nevoid basal cell carcinoma syndrome have a high potential for recurrence (60%). [6] In rare instances, they may be associated with development of ameloblastoma and squamous cell carcinoma. [2]
Follow-up should involve annual conventional radiographic surveillance preferably supplemented with cone-beam computed tomography or magnetic resonance imaging (MRI) when there is suspicion of recurrence.
Rib anomalies are reported in 30−60% of patients with this condition, of which bifid ribs are more common, but these were not seen in our case. [2]
Calcification of falx cerebri is one of the most frequent radiological features, seen in 37−79% of cases with nevoid basal cell carcinoma syndrome. [1] Kimonis et al. reported calcification of falx cerebri to be more common in patients older than 20 years. [4]
Ovarian fibromas and cysts are seen in 25−50% of female patients with this syndrome and are often bilateral (75%). They do not reduce fertility but may undergo torsion (twist). [2]
Medulloblastomas (now termed primitive neuroectodermal tumour) are seen in 3−5% of patients with nevoid basal cell carcinoma syndrome, usually within the first 2 years of life. Molecular genetic studies for PTCH1 gene mutations may be helpful in early diagnosis in such cases. [1],[2] Neurological examination, including MRI, is recommended every 6 months until 3 years of age followed by annual follow-up until 7 years of age, after which medulloblastoma is very unlikely. [2]
Our patient was older than the risk group for occurrence of medulloblastoma and did not have any signs or symptoms of the disease. The patient is, however, at risk of developing intracranial tumors such as meningioma in the future, and hence, neurological follow-up is recommended. [2]
Once the diagnosis of nevoid basal cell carcinoma syndrome is made, screening must be carried out in other family members and genetic counselling must be offered. This syndrome is usually a hereditary condition with germ line mutations in the PTCH1 gene reported in members of the same family in 50% of the cases. [1]
| 1. |
De Ortega G, Amezaga A, García Arregui O, Zepeda Nuño S, Acha Sagredo A, Aguirre Urizar JM, et al. Gorlin-Goltz syndrome: Clinicopathologic aspects. Med Oral Patol Oral Cir Bucal 2008;13:E338-43.
[Google Scholar]
|
| 2. |
Lo Muzio L. Nevoid basal cell carcinoma syndrome (Gorlin Syndrome). Orphanet J Rare Dis 2008;3:32.
[Google Scholar]
|
| 3. |
Evans DGR, Ladusans EJ, Rimmer S, Burnell LD, Thakker N, Farndon PA. Complication of nevoid basal cell carcinoma syndrome: Results of a population based study. J Med Genet 1993;30:460-4.
[Google Scholar]
|
| 4. |
Kimonis VE, Goldstein AM, Pastakia B, Yang ML, Kase R, Digiovanna JJ, et al. Clinical manifestations in 105 persons with Nevoid basal cell carcinoma syndrome. Am J Med Genet 1997;69:299-308.
[Google Scholar]
|
| 5. |
Parikh NR. Nonsyndromic multiple odontogenic keratocysts: Report of a case. Adv Dent Res 2010;1:71-4.
[Google Scholar]
|
| 6. |
Macdonald-Jankowski DS. Keratocystic odontogenic tumour: Systematic review. Dentomaxillofac Radiol 2011;40:1-23.
[Google Scholar]
|
Fulltext Views
5,352
PDF downloads
2,059





![[Figure - 1]](#fig_ijdvl_2015_81_4_414_159945_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2015_81_4_414_159945_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2015_81_4_414_159945_f3.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2015_81_4_414_159945_t4.jpg){kind=link}