
Translate this page into:
Non-healing leg ulcers in HbE-beta thalassemia

Corresponding author: Dr. Vinay Keshavamurthy, Department of Dermatology, Venereology and Leprology, Post Graduate Institute of Medical Education and Research, Chandigarh, India. vinay.keshavmurthy@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Sharma A, Keshavamurthy V. Non-healing leg ulcers in HbE-beta thalassemia. Indian J Dermatol Venereol Leprol. 2025;91:266-7. doi: 10.25259/IJDVL_154_2024
A 26-year-old man presented for evaluation of painful, chronic, non-healing leg ulcers of 12 years’ duration. On cutaneous examination, there were three well-defined, irregularly marginated tender ulcers present on the medial and lateral aspect of the lower third of the leg overlying the malleoli [Figure 1]. The largest of the ulcers measured 14×10 cm, had focally undermined edges. The floor was covered with yellowish to brownish necrotic slough [Figure 2]. Surrounding skin showed the presence of brownish hyperpigmentation and slight induration (reflective of underlying fibrosis). There was associated restricted movement of both ankle joints. On abdominal examination, the spleen was palpable 6 cm below the left costal margin. Venous and arterial Doppler were normal. Skin biopsy showed partially ulcerated stratified squamous epithelium lined by fibrinous exudate. The rest of the fragments showed hyperkeratosis, parakeratosis, and spongiosis in the upper dermis, the presence of focal vessel wall damage, neutrophilic inflammation, and karyorrhectic debris along with areas of fibrinoid necrosis. There was evidence of an old bleed in the vicinity of vessels in the form of haemosiderin laden macrophages. There was also dense mixed inflammation comprising lymphocytes, plasma cells, histiocytes, neutrophils and a few eosinophils. Haematological investigations revealed severe anaemia (Hb- 6.2 g/dL), raised reticulocyte count (3.2%), low haptoglobin (23.7 mg/dL); high performance liquid chromatography (HPLC) revealed HbA2 fraction of 61.1% (reference range 2.1–3.5) and HbF fraction 31.1% (reference range 0.2–1.0). The phenotype of the disease was consistent with thalassemia major (transfusion-dependent beta thalassemia). Due to the considerably high fraction of HbA2, the genotype was consistent with a diagnosis of a double heterozygous state for HbE with beta thalassemia (HbE-beta thalassemia). The patient was subsequently treated with blood transfusion, daily dressing, oral antibiotics based on culture sensitivity, folic acid 5 mg per day and hydroxyurea 500 mg per day after haematological consultation. He remains under follow-up. For further management, he was planned for splenectomy by haematology.
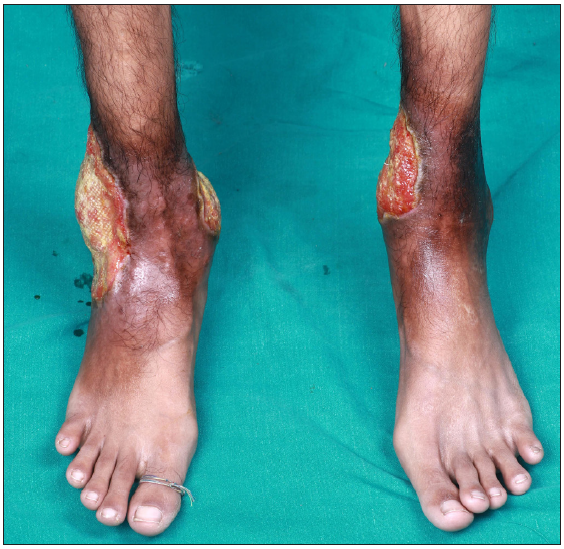
- Three well-defined, irregularly marginated ulcers, the largest of size 14×10 cm on the medial and lateral malleolar aspect.
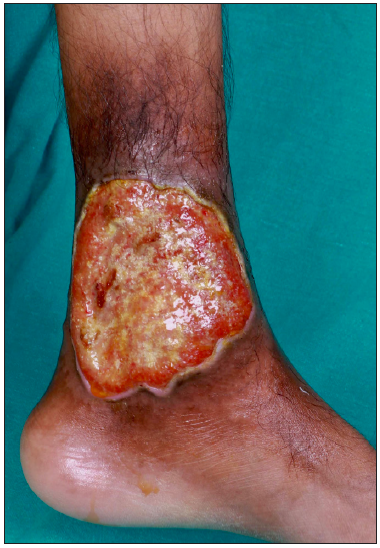
- The largest ulcer with focally undermined edges and floor covered with yellowish to brownish necrotic slough with serosanguinous discharge and perilesional hyperpigmentation.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.




