
Translate this page into:
Omalizumab and dupilumab for the treatment of autosomal-recessive DOCK8 hyper-IgE syndrome
Corresponding author: Dr. Youkun Lin, Department of Dermatology and Venereology, The First Affiliated Hospital of Guangxi Medical University, Nanning-530022, Guangxi, China. linyoukun7@aliyun.com
-
Received: ,
Accepted: ,
How to cite this article: Guo T, Wei L, Karki S, Wen S, Li Q, Lin Y. Omalizumab and dupilumab for the treatment of autosomal-recessive DOCK8 hyper-IgE syndrome. Indian J Dermatol Venereol Leprol. 2025;91:94-6. doi: 10.25259/IJDVL_348_2023
Dear Editor,
We present an 11-year-old boy with autosomal-recessive DOCK8 hyper-IgE syndrome who underwent treatment with both omalizumab and dupilumab. The patient had a recurrent oral mass for 5 years and atopic dermatitis for 2 years, despite treatment with topical and systemic steroids and oral antihistamines. He also gave history of recurrent pneumonia and abdominal abscess. Physical examination revealed a granulomatous mass in the right buccal mucosa, accompanied by missing teeth, widespread erythematous papules, xerosis, and hyperpigmentation [Figures 1a, 1b and 1c]. A skin biopsy confirmed widespread dermal granulomas composed of histiocytes, lymphocytes, and eosinophilic granulocytes in the superficial and mid dermis. [Figures 1d and 1e]. Genetic analysis indicated a homozygous deletion in exons 2–11 of the DOCK8 gene, confirming the diagnosis of autosomal-recessive hyper-IgE syndrome with elevated serum IgE levels at 7591.2 IU/mL (normal range: 0–100 IU/mL).
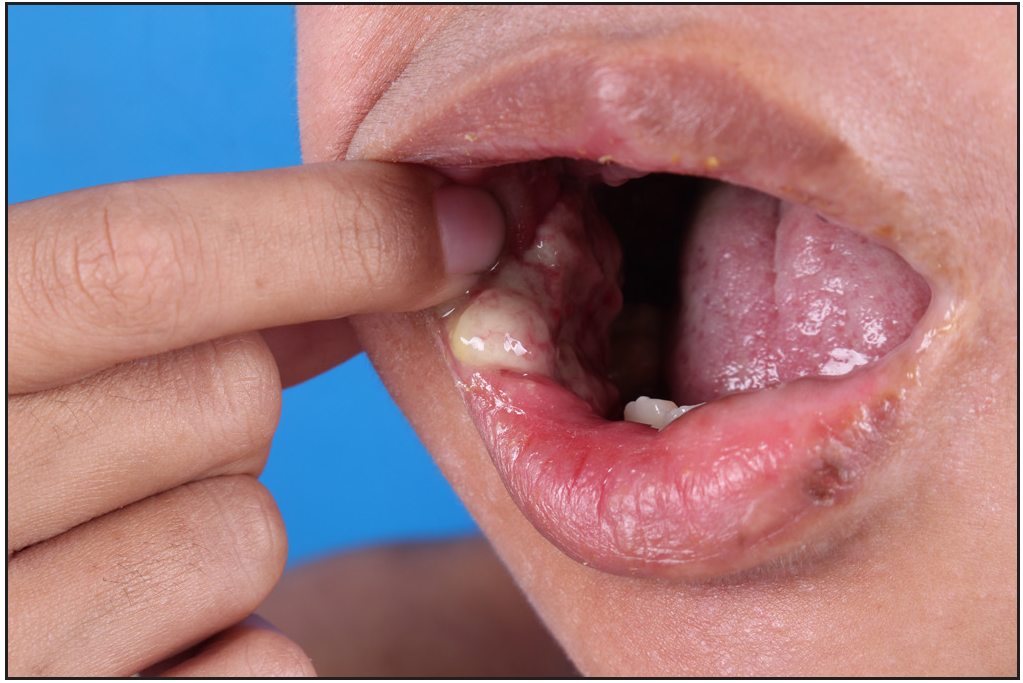
- Granulomatosis-like mass in the right buccal mucosa before treatment.
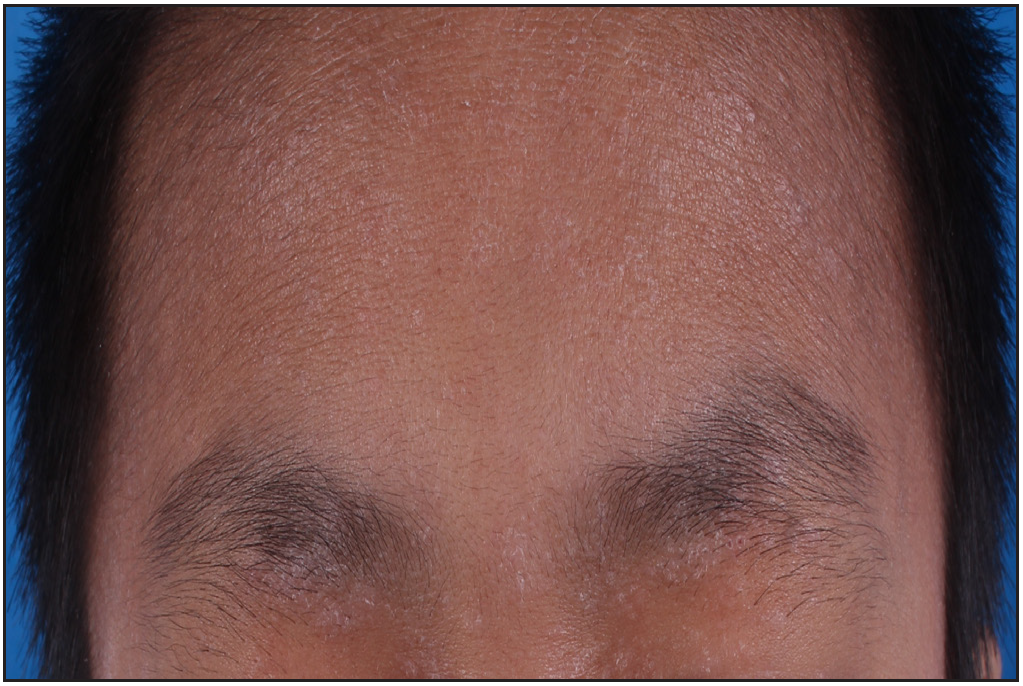
- Erythema and xerosis on the forehead and eyelids before treatment.
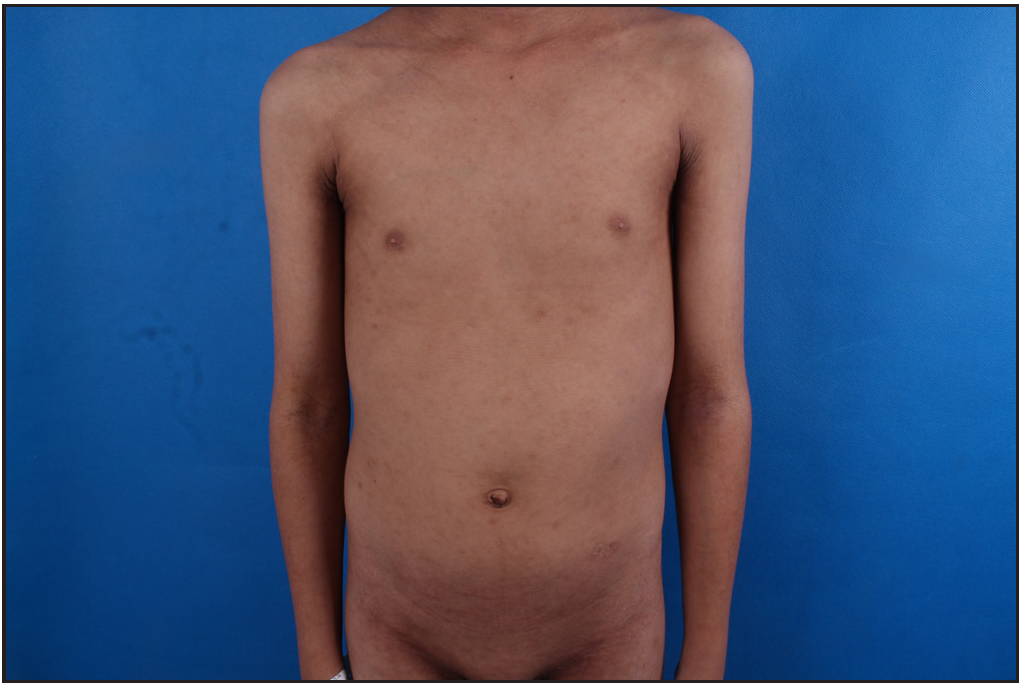
- Erythema and xerosis on the trunk and upper limbs before treatment.
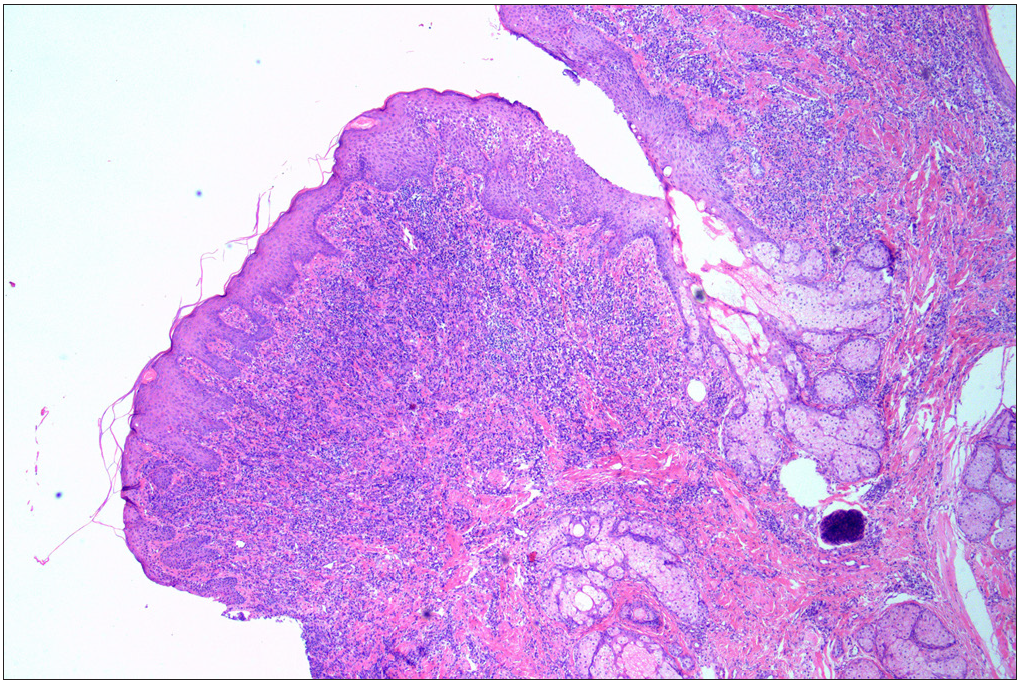
- A skin biopsy from the oral nodule showing significant infiltration of histocytes, lymphocytes, and eosinophilic granulocytes in the superficial mid-layer of the dermis, consistent with cutaneous granulomatosis (Haematoxylin and eosin, original magnification × 40).
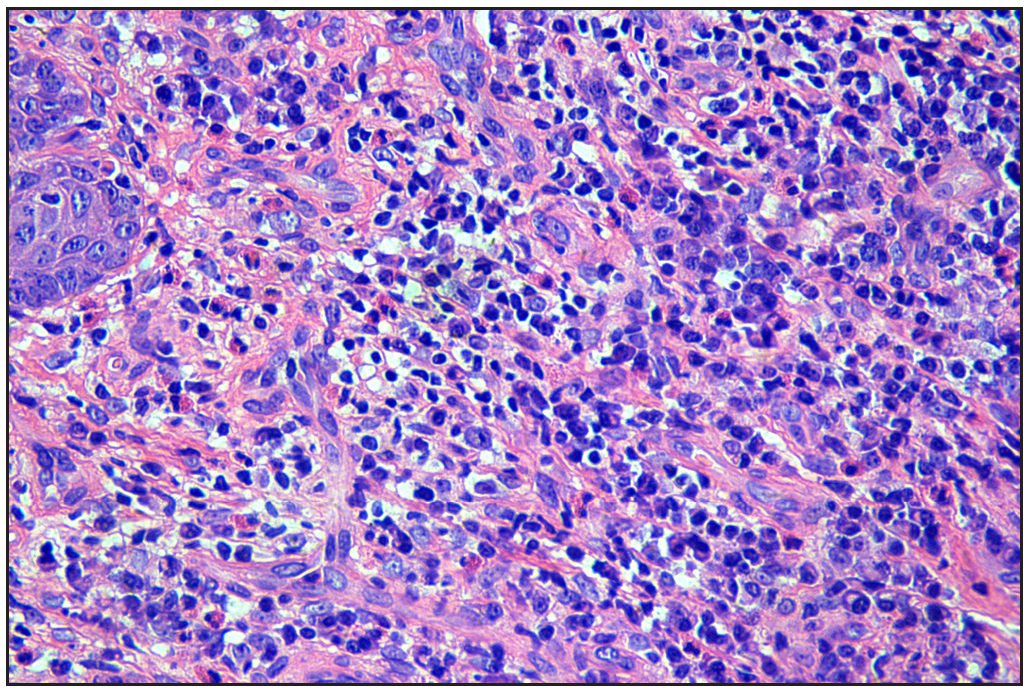
- A skin biopsy from the oral nodule showing a significant infiltration of histocytes, lymphocytes, and eosinophilic granulocytes in the superficial mid-layer of the dermis, consistent with cutaneous granulomatosis (Haematoxylin and eosin, original magnification × 400).
Despite intensive care and preventive anti-infective measures, patient’s symptoms persisted. Omalizumab was administered (300 mg subcutaneously every 4 weeks) on August 25, 2021, and September 26, 2021. However, there was no significant symptomatic improvement or reduction in IgE levels until November 9, 2021 (7049.5 IU/mL). Given the patient’s severe symptoms, the fact that the eczematous lesions improved almost two months after initiating omalizumab,1 and the family’s request for alternate treatment, the patient was switched to dupilumab on November 10, 2021 (300 mg subcutaneously every 4 weeks). After 8 weeks of dupilumab treatment, we observed significant improvement in atopic dermatitis and complete resolution of the granulomatous mass. [Figures 2a, 2b and 2c]. IgE levels reduced to 161.5 IU/mL. Monthly dupilumab injections effectively controlled and stabilized the symptoms until October 9, 2022, followed by subsequent recurrence of the oral granulomatosis-like mass when the injection interval was extended to 1.5 months. The patient has remained recurrence-free since returning to monthly injections.
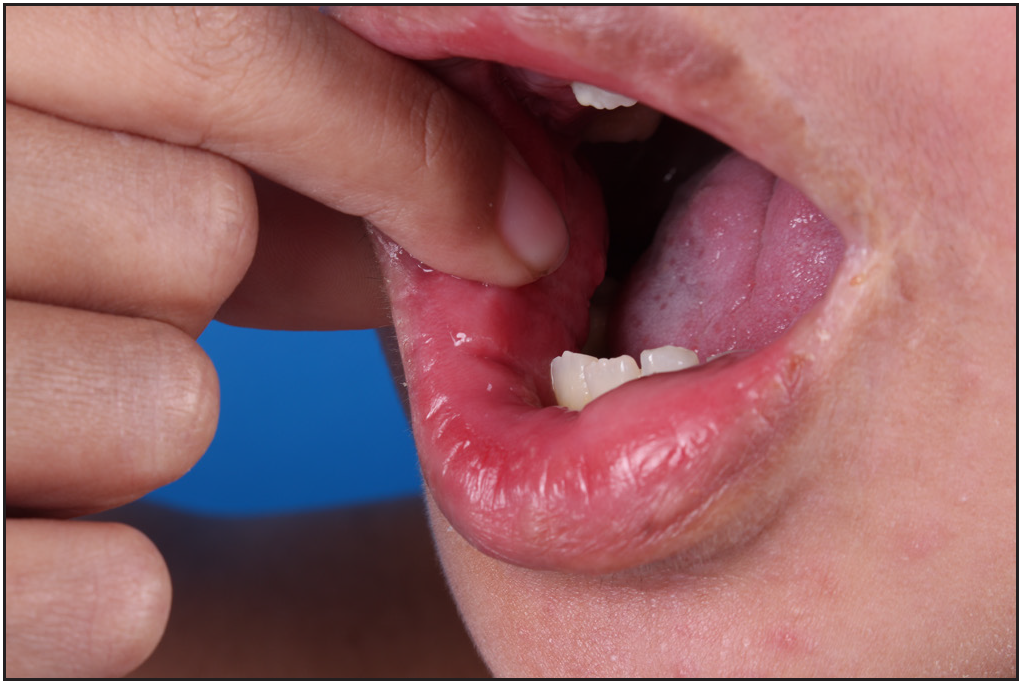
- Granulomatosis-like mass in the right buccal mucosa disappeared after 8 weeks of dupilumab therapy.
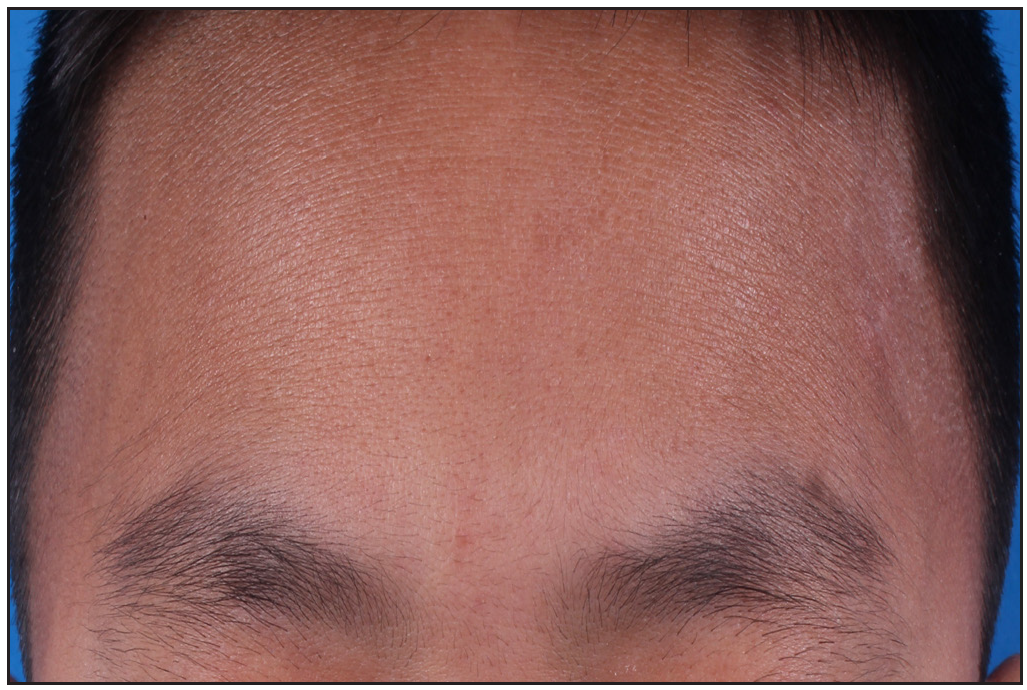
- Erythema and xerosis on the forehead and eyelids disappeared after 8 weeks of dupilumab therapy.
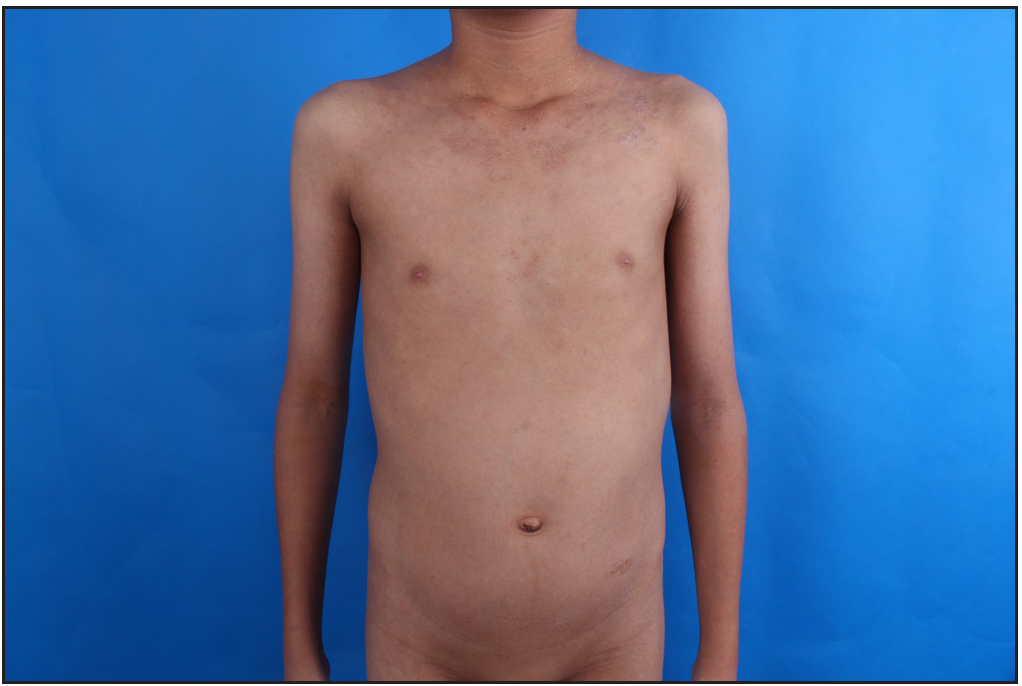
- Erythema and xerosis on the trunk and upper limbs disappeared after 8 weeks of dupilumab therapy.
Autosomal-recessive hyper-IgE syndrome is a rare immunodeficiency disorder resulting from DOCK8 defects. It is characterised by recurrent high serum IgE, refractory atopic dermatitis, cutaneous and respiratory infections, and an increased risk of early malignant tumours.2 While there is no established treatment paradigm for hyper-IgE syndrome, biological formulations like omalizumab and dupilumab have shown potential for alleviating symptoms.2–4.
Omalizumab, a humanized recombinant anti-IgE antibody, is beneficial in patients with hyper-IgE syndrome.4 However, the present patient used it for less than three months, precluding a comprehensive assessment of the drug’s response. In contrast, dupilumab, a fully humanised IgG4 monoclonal antibody, impedes downstream signalling of IL-4/13 cytokines by binding to the alpha-subunit of the IL-4R and IL-13R, leading to reduced IgE production and relief from Th2 cell-mediated disease.3 Substantial evidence suggests that Th2-mediated IL-4/13 signals play a role in autosomal-recessive hyper-IgE syndrome.2 Successful treatment of the present patient with dupilumab further bolsters this perspective.
We further observed that dupilumab was effective in decreasing the granulomatosis in the present patient. This suggests that dupilumab may enhance the host’s ability to resist pathogenic microbiological infections by immune response regulation. However, further studies are needed to fully comprehend the underlying mechanism.
Limitation of the present report is the short washout period between omalizumab and dupilumab. The half-life of serum elimination for omalizumab is 24 days, and clinical trials typically necessitate a washout period of four half-lives. However, in this case, such a prolonged washout period was not feasible.
In summary, the patient’s symptoms improved and his recurrence was arrested after switching to dupilumab, suggesting its effectiveness in hyper IgE syndrome.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of Artificial Intelligence (AI)-Assisted Technology for manuscript preparation
The authors confirm that there was no use of Artificial Intelligence (AI)-Assisted Technology for assisting in the writing or editing of the manuscript and no images were manipulated using the AI.
References
- Role of omalizumab in a patient with hyper-IgE syndrome and review dermatologic manifestations. Asian Pac J Allergy Immunol. 2009;27:233-6.
- [PubMed] [Google Scholar]
- Treatment options for DOCK8 deficiency-related severe dermatitis. J Dermatol. 2021;48:1386-93.
- [CrossRef] [PubMed] [Google Scholar]
- Clearance of atypical cutaneous manifestations of hyper-IgE syndrome with dupilumab. Pediatr Dermatol. 2022;39:940-2.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Omalizumab for STAT3 hyper-IgE syndromes in adulthood: A case report and literature review. Front Med (Lausanne). 2022;9:835257.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




