
Translate this page into:
Pathogenic variants in PSENEN and NCSTN genes cause ‘follicular’ Dowling-Degos disease: Report of five unrelated Indian families
Corresponding author: Dr. Sujay Khandpur, Department of Dermatology and Venereology, All India Institute of Medical Sciences, New Delhi, India. sujay_khandpur@yahoo.com
-
Received: ,
Accepted: ,
How to cite this article: Gupta V, Khandpur S, Bhalla D, Sarathe C, Singh S, Yenamandra VK. Pathogenic variants in PSENEN and NCSTN genes cause ‘follicular’ Dowling-Degos disease: Report of five unrelated Indian families. Indian J Dermatol Venereol Leprol. doi: 10.25259/IJDVL_1767_2024
Abstract
Background
Follicular Dowling-Degos disease (DDD) is a rare clinically and histologically distinct genodermatosis. However, its genetic basis has not been well-studied.
Objective
To describe the clinical, histological, and mutational spectrum of follicular DDD in 10 patients from five unrelated Indian families.
Methods
Clinical and histological features of patients were recorded. Whole exome sequencing was done on the venous blood of probands and their family members, and its results were validated by Sanger sequencing.
Results
All patients presented with open comedones, small follicular keratotic papules, and fine pitted and shallow crateriform scars predominantly on the face, back and flexural sites. The skin biopsy showed follicular plugs along with downward elongation and branching of pigmented rete ridges confined to the follicular infundibulum. Whole exome sequencing revealed distinct pathogenic variants in the PSENEN (3 families) and NCSTN (2 families) genes of probands and their affected family members, as validated by Sanger sequencing.
Limitations
Functional significance of the gene mutations in disease pathogenesis could not be assessed by cell culture studies and knock-down experiments.
Conclusion
Our study identified PSENEN and NCSTN gene mutations as the genetic basis of follicular DDD. These genes encode proteins involved in the Notch signalling pathway and can potentially explain the predominantly folliculocentric phenotype of follicular DDD.
Keywords
Dowling degos disease
Follicular
Genetics
PSENEN
NCSTN

Introduction
Dowling-Degos disease (DDD) is a rare genodermatosis with flexural reticulate hyperpigmentation caused by pathogenic variants in KRT5 (intermediate filament), POFUT1 or POGLUT1 (components of the Notch-signalling pathway) genes.1 Recently, pathogenic variants in PSENEN and NCSTN genes (components of the γ-secretase complex) have been implicated in DDD pathogenesis, particularly in patients with associated hidradenitis suppurativa.2-5 Our group has previously described a follicular variant of DDD as a distinct entity. It is clinically characterised by pits, comedones, and cysts on the face, trunk, and flexures, and histopathologically by thin, elongated, and branched pigmented rete ridges restricted to hair follicles.6 Here, we describe the mutational spectrum of our patients with follicular DDD.
Methods
This was a case series including 10 patients with follicular DDD from five unrelated Indian families. Clinical findings were recorded, and photographs of skin lesions were taken. A 4mm skin punch biopsy was done from representative skin lesions of consenting patients. Post informed consent, whole exome sequencing was performed as described previously.7 Briefly, gDNA was isolated from the peripheral blood of all index patients and subjected to exome library preparation and paired-end sequencing (at >100x coverage) as per the manufacturer’s recommendations (Illumina Inc., USA). Raw sequencing reads were quality trimmed and aligned to the human reference genome (GRCh38), followed by variant calling using the DRAGENTM pipeline (Illumina Inc., USA). Finally, variant annotation and prioritisation were performed based on the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) guidelines.8
Results
Five unrelated non-consanguineous Indian families with 10 patients (7 males and 3 females) affected with follicular DDD were studied. Clinically, all patients had striking features of follicular occlusion like open comedones, small follicular keratotic papules, and fine-pitted shallow crateriform scars, predominantly on the face, back, and flexures, that were noticed first during adolescence [Figures 1a and 1b]. Additionally, five affected patients from three families (AII.2, BII.1, and DII.1-3) had brownish hyperpigmented macules on the flexures, typical of classical DDD, while another patient (AI.1) had discrete hypopigmented macules on the back and lower limbs. The clinical findings of the patients are summarised in Table 1. None of the patients or family members has developed hidradenitis suppurativa to date, with a follow-up duration of 4-6 years.
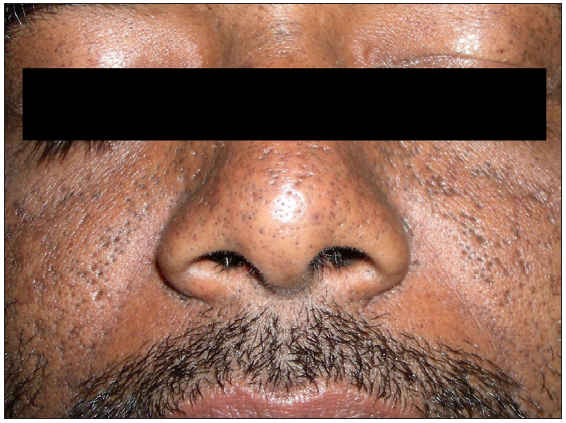
- Crateriform pits and open comedones on the face. Note the hyperpigmented macules of classic DDD on the upper eyelids.
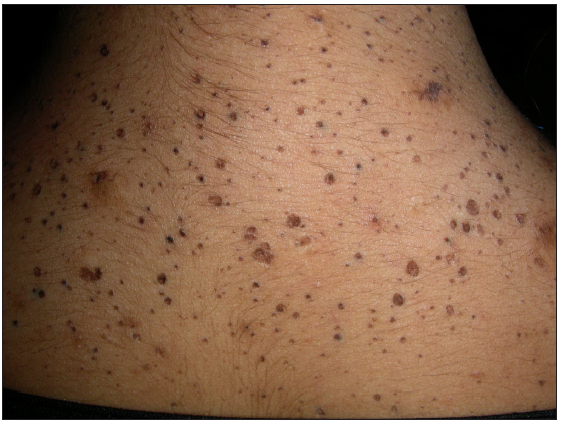
- Crateriform pits and open comedones on the upper back.
| Family Patient | AI.1 | AII.2 | BI.1 | BII.1 | CII.1 | DII.1 | DII.2 | DII.3 | EI.1 | EII.3 |
|---|---|---|---|---|---|---|---|---|---|---|
| Age (years) | 50 | 20 | 48 | 16 | 21 | 30 | 23 | 19 | 52 | 19 |
| Gender | M | F | M | F | F | M | M | M | M | M |
| Age of onset in years | NA | 9 | NA | 11 | 13 | 12 | 8 | 11 | NA | 14 |
| Open comedones | + | + | + | + | + | + | + | + | + | + |
| Follicular keratotic papules | + | + | + | + | + | + | + | + | + | + |
| Pitted and crateriform scars | + | + | + | + | + | + | + | + | + | + |
| Sites of papules, comedones and scars | Face, upper back | Face, upper back, cubital fossae | Face, back, cubital fossae, axillae | Face, back, axillae | Face, upper chest, back, cubital and popliteal fossae, arms and thighs | Face, back, cubital fossae, axillae, legs | Face, upper back, cubital and popliteal fossae, axillae, distal forearms | Face, upper back, popliteal ossae, axillae, distal forearms | Face, back | Face, back |
| Mottled hyperpigmentation | - | + (flexures; axillae, groins, cubital fossae) | - | + (flexures; axillae, groins, cubital fossae) | - | + (ears) | + (flexures > generalized) | ±flexures; axillae, groins, cubital fossae) | - | - |
| Hypopigmented macules | + (shoulders, lower limbs) | - | - | - | - | - | - | - | - | - |
| Genotype | NCSTN, likely-pathogenic variant, c.l493T>C, pXeu498Pro | PSENEN, pathogenic variant, c.l94T>G, p.Leu65Arg | PSENEN, pathogenic variant, c.62- ldelG | PSENEN. likely-pathogenic variant, c.65G>A, p.Gly22Glu | NCSTN, pathogenic variant c.1060_61 insTGCAGTTA, p.Val354fs | |||||
M: Male, F: Female, NA: Not applicable
Skin punch biopsy of an open comedone or follicular keratotic papule, done in eight patients, showed similar histological features: keratinous follicular plugs with downward elongation and branching of pigmented rete ridges originating from the follicular infundibulum, with non-involvement of the interfollicular epidermis [Figure 2].
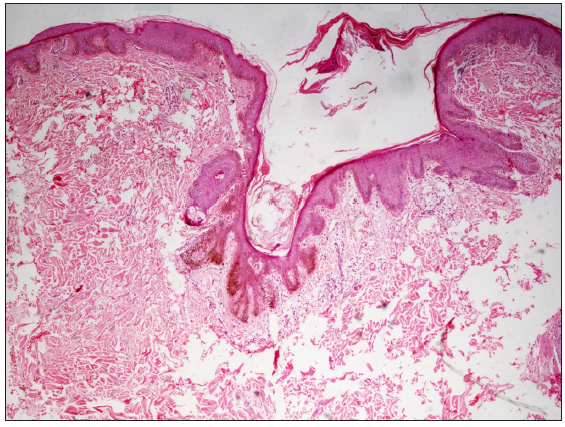
- Histopathology showing downward slender elongation of pigmented rete pegs confined to the follicular epithelium (Haematoxylin and eosin, 400x).
We found three distinct variants in the PSENEN gene, one novel (Family D: heterozygous missense mutation in the exon3, c.65G>A, p.Gly22Glu, likely-pathogenic) and two previously reported (Family B: heterozygous missense mutation in exon 4, c.194T>G, p.Leu65Arg, pathogenic and Family C: splice site mutation in exon 3, c.62-1delG, pathogenic). Another two novel variants were identified in the NCSTN gene (Family A: heterozygous missense mutation in exon 13, c.1493T>C, p.Leu498Pro, likely-pathogenic and Family E: heterozygous frameshift mutation in exon 9, c.1060_61insTGCAGTTA, p.Val354fs, pathogenic). These were not present in the publicly available global (https://gnomad.broadinstitute.org/) and Indian population (https://clingen.igib.res.in/indigen/) genome databases, and all missense variants were predicted to be deleterious by in-silico pathogenicity prediction tools. No additional variants were identified in any other genes implicated in DDD (KRT5, POFUT1, POGLUT1). Sanger sequencing revealed that all identified variants co-segregate in only the affected family members, confirming an autosomal dominant inheritance [Figure 3].
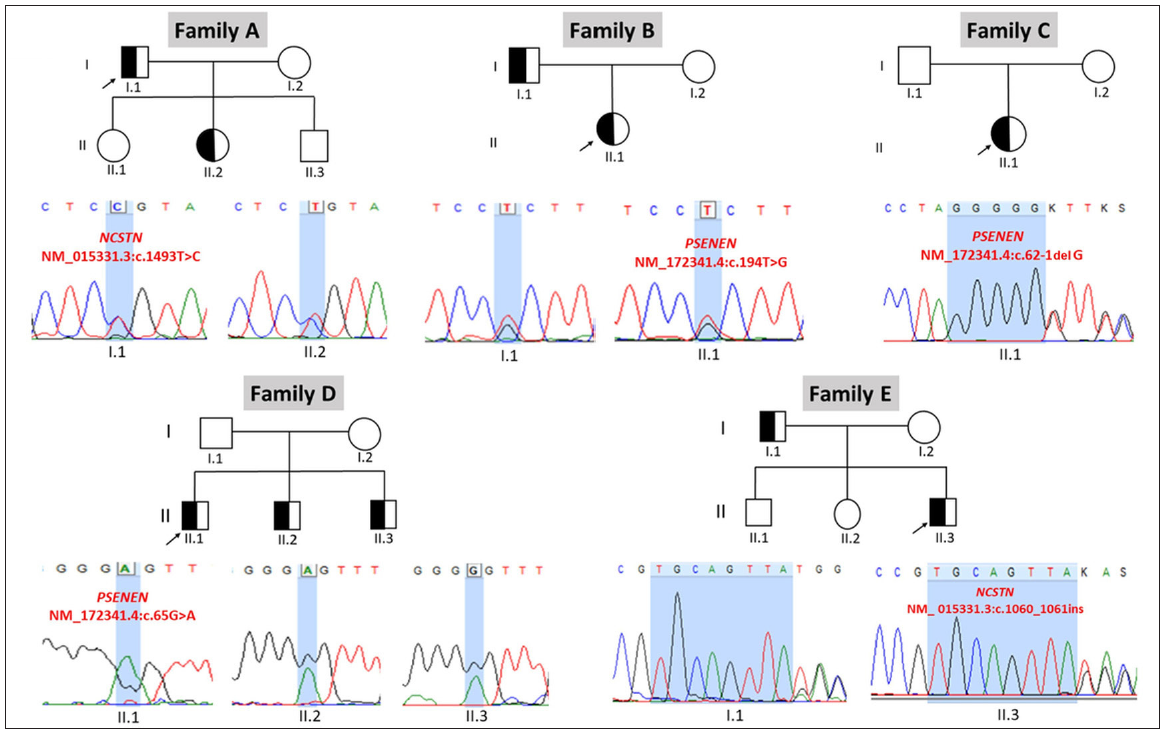
- Sequence chromatograms depicting the identified pathogenic or likely pathogenic variants in PSENEN and NCSTN genes in the affected follicular DDD patients and their family members (arrows indicate proband).
Discussion
Over the years, several variants of DDD have been described, including a generalised variant, Galli-Galli disease, Haber’s syndrome, and overlap with the reticulate acropigmentation of Kitamura. There are several reports of patients with DDD having additional comedo-like papules, epidermoid cysts, and hidradenitis suppurativa.1 In 2013, our group reported a distinct clinical and histological variant, follicular DDD.6 The clinical features include follicular keratotic (comedo-like) papules and pitted scars on the face, trunk, and flexures. In some, these lesions may be accompanied by pigmentary changes of classical DDD. Histopathology of comedo-like papules or pitted scars show typical features of DDD - elongated and branching rete ridges with increased basilar melanisation - but restricted to the epithelium of a dilated hair follicle plugged with keratin.6 Since then, a few more reports of this DDD variant have emerged in the literature; however, its genetic basis has not been well studied.
In this report, we found pathogenic variants in PSENEN (3 families) and NCSTN genes (2 families) in our patients with follicular DDD. It is noteworthy that mutations in both PSENEN and NCSTN genes have been reported in patients with familial hidradenitis suppurativa9,10 or DDD-hidradenitis suppurativa overlap,2-5 providing a causal link between two clinically contrasting diseases. Most reported mutations in the PSENEN gene causing DDD phenotype, with or without hidradenitis suppurativa, have affected exons 3 and 4, similar to our families (families C D, exon 3; family B, exon 4).2-5 Occurrences of different mutations at the same site may suggest that it may be a mutation hotspot. Interestingly, six of 10 patients from three families in our cohort had co-existent pigmentary changes of classic DDD as well. The variation in clinical features within the same family, particularly in the presence and extent of pigmentary change, points towards the variable phenotypic expression of the same genetic mutation.
None of our patients had developed hidradenitis suppurativa till the last follow-up, though one could argue that the clinical phenotype (with open comedones, keratotic papules, and pitted scars) is a forme fruste of follicular occlusion syndrome. Notably, frameshift mutations in the PSENEN gene have been reported in familial comedone syndrome.11 However, histopathology of the comedo or follicular keratotic papules in our series showed elongated and branched pigmented rete ridges affecting the follicular infundibulum. This pattern is characteristic of DDD and has not been described in hidradenitis suppurativa, familial comedone syndrome, or other follicular pathologies. Comedonal Dariers disease12 and comedogenic lupus erythematosus13 can also be considered in the differential diagnoses of such a clinical presentation. However, in such a case, patients usually have other characteristic skin lesions as well, and histopathology shows distinct features.
Mutations in PSENEN and NCSTN genes result in haploinsufficiency of the γ-secretase complex, which is necessary for the activation of the Notch signalling pathway.3 Aberrations in the Notch pathway can result in features of follicular occlusion and pigmentary changes. Particularly relevant to the pigmentary anomaly, a PSENEN knock-down zebrafish model provides evidence for disordered migration and differentiation of pigment cells, resulting in scattered pigmentation like in DDD.2
Limitation
The functional significance of PSENEN and NCSTN gene mutations in disease pathogenesis could not be assessed by cell culture studies and knock-down experiments.
Conclusion
We found mutations in the PSENEN and NCSTN genes that encode protein components of the γ-secretase complex as the genetic basis of follicular DDD. Our observations lend support to the shared folliculocentric theory between follicular DDD and hidradenitis suppurativa. Studying the functional significance of the PSENEN and NCSTN mutations can provide further insights into disease pathogenesis and identify therapeutic targets.
Ethical approval
Institutional Review Board approval is not required as this case-series is a review of cases seen at our institute over a period of more than 5 years.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
VKY acknowledges funding support from the Council of Scientific and Industrial Research (CSIR), through grant no. MLP2001 and OLP1164.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Dowling-Degos disease: A review. Int J Dermatol. 2021;60:944-50.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in γ-secretase subunit-encoding PSENEN underlie dowling-Degos disease associated with acne inversa. J Clin Invest. 2017;127:1485-90.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- A phenotype combining hidradenitis suppurativa with dowling-Degos disease caused by a founder mutation in PSENEN. Br J Dermatol. 2018;178:502-8.
- [CrossRef] [PubMed] [Google Scholar]
- Novel nicastrin mutation in hidradenitis suppurativa-Dowling-Degos disease clinical phenotype: More than just clinical overlap? Br J Dermatol. 2020;183:758-9.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- A loss-of-function NCSTN mutation associated with familial dowling degos disease and hidradenitis suppurativa. Exp Dermatol. 2023;32:1935-45.
- [CrossRef] [PubMed] [Google Scholar]
- Follicular dowling degos disease: A rare variant of an evolving dermatosis. Indian J Dermatol Venereol Leprol. 2013;79:802-4.
- [CrossRef] [PubMed] [Google Scholar]
- Application of whole exome sequencing in elucidating the phenotype and genotype spectrum of junctional epidermolysis bullosa: A preliminary experience of a tertiary care centre in India. J Dermatol Sci. 2017;86:30-6.
- [CrossRef] [PubMed] [Google Scholar]
- Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Insights from γ-Secretase: Functional genetics of hidradenitis suppurativa. J Invest Dermatol. 2021;141:1888-96.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Gamma-secretase gene mutations in familial acne inversa. Science. 2010;330:1065.
- [CrossRef] [PubMed] [Google Scholar]
- A frameshift mutation in PEN-2 causes familial comedones syndrome. Dermatology. 2015;231:77-81.
- [CrossRef] [PubMed] [Google Scholar]
- Comedogenic lupus: A rare variant of chronic cutaneous lupus erythematosus - case series. An Bras Dermatol. 2023;98:159-67.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




