Translate this page into:
Piebaldism with non-intertriginous freckles: What does it mean?
Correspondence Address:
Nilendu Sarma
PN Colony, Sapui Para, Bally, Howrah - 711 227, West Bengal
India
| How to cite this article: Sarma N, Chakraborty S, Bhanja DC, Bhattachraya SR. Piebaldism with non-intertriginous freckles: What does it mean?. Indian J Dermatol Venereol Leprol 2014;80:163-165 |
Sir,
Piebaldism is a rare autosomal dominant condition characterized by the presence of depigmented patches that are congenital, small to large, sharply demarcated, roughly symmetric and distributed over the frontal scalp, forehead, ventral chest, abdomen and mid-portion of extremities. A white forelock is present in most patients with or without leukotrichia of other areas. Some patients also have hyperpigmented macules. On biopsy, the depigmented macule lacks melanocytes while the hyperpigmented macules have a normal number of melanocytes which may show an aberrant morphology. Two cases of piebaldism in a Hindu family involving the father and daughter are reported here.
The daughter was a 16-year-old girl who had a white forelock on the frontal scalp, patches of white hair on the bilateral eyebrows [Figure - 1] and a large depigmented patch on the extensor surface over the right knee. The extent of the patch could be well-recognized despite not having a well-defined margin all around. The area was not homogenously affected; depigmented patches intermingled with normally pigmented skin. In addition, there were numerous smaller hyperpigmented macules [Figure - 2]. These pigmentary changes were present since birth.
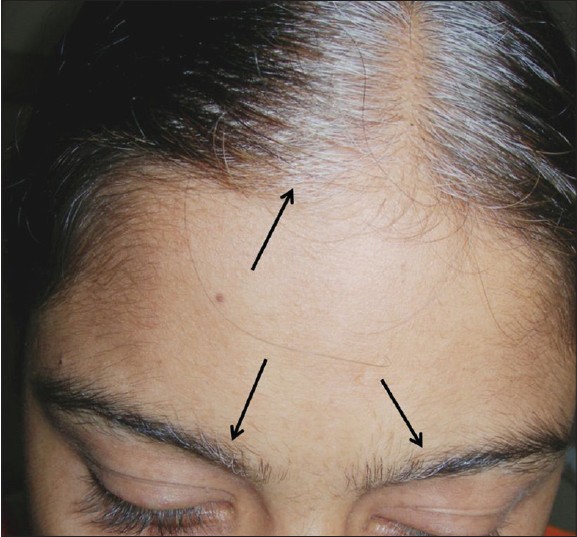 |
| Figure 1: Localised areas of white hair involving frontal scalp and both eyebrows since birth |
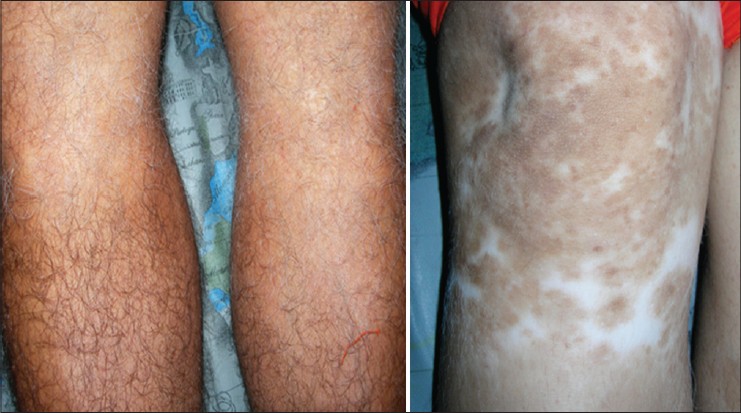 |
| Figure 2: Hypopigmented patch with white hair on both legs just below knee in the father (left panel) and a depigmented macule on the right knee in the daughter having white hair and numerous hyerpigmented macules within it |
Her father had symmetrically distributed ill-defined areas of hypopigmentation with overlying white hair on the medial aspect of both knees since birth. There were a few islands of depigmented skin present in the hypopigmented area [Figure - 2]. He had a congenital white forelock on the frontal scalp. He told us that the area under the white forelock was also hypopigmented at birth but repigmented later. At the time of examination, no background hypopigmentation was noted.
Both father and daughter had numerous freckle-like macules on the limbs since birth these were most dense on the distal parts [Figure - 3] since birth. The girl had a few (<5) café-au-lait macules on the distal forearm and lower legs. Both of them were of normal intelligence and had not suffered any major illnesses. Neither of them had any neurofibromas or Lisch nodules and they did not have any systemic abnormalities.
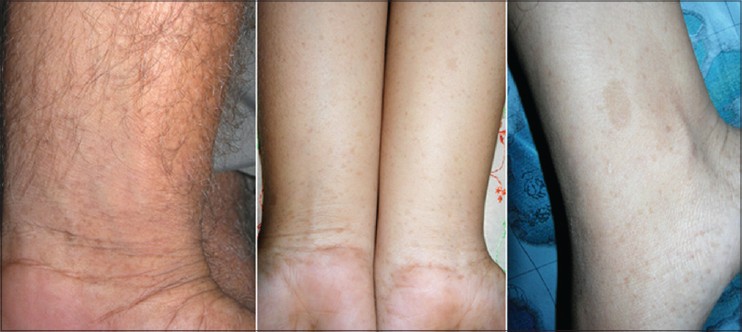 |
| Figure 3: Left panel shows numerous freckles on the forearm in the father and right panel shows freckles and a Café-an-lait macule on the legs and forearm of the daughter |
There was no history of consanguineous marriages in the family and no other relatives had a similar illness [Figure - 4].
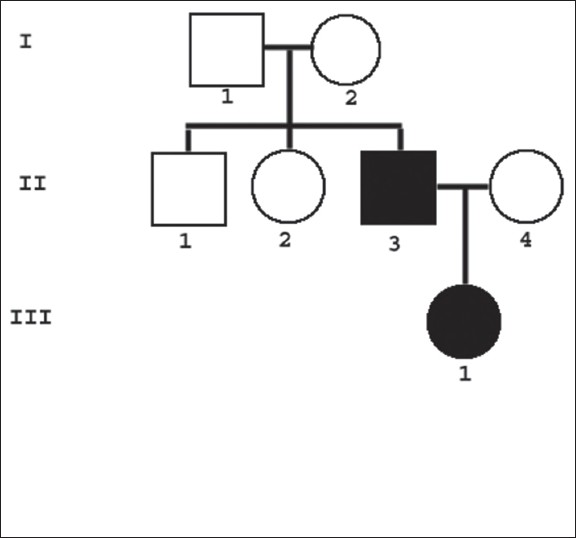 |
| Figure 4: A three generation pedigree chart of the family depicting the affected members |
Histopathology from depigmented skin in the girl showed absence of any visible melanocytes in otherwise normal appearing skin. Histopathology of the freckles showed a pigmented basal layer without any elongation in rete ridges or increased number of basal melanocytes.
Based on clinical appearance, the cases were diagnosed as piebaldism.
Piebaldism results from different mutations including many deletions generally involving c-KIT proto-oncogene and occasionally SLUG gene, a zinc-finger neural crest transcription factor or some other unidentified gene [1] leading to a defect in the transmembrane tyrosine kinase receptor that in turn results in abnormal proliferation, survival or distribution of melanoblasts and melanocytes during early embryologic development. Thus, piebaldism is primarily characterized by depigmentation and the extent of depigmentation is generally used to grade the clinical severity. Four different classes of KIT mutations are now known based on severity and types of mutation in the gene. Class one KIT gene mutation consists of missense substitution and is characterized by the most severe phenotype with depigmentation that even reaches the dorsal surfaces of head and trunk. [2] Milder hypopigmented lesions may improve with age. [3] The father showed regression of hypopigmentation on the frontal scalp but the white forelock persisted.
Not uncommonly, patients with piebaldism may also develop three types of hyperpigmentation. [4] These are hyperpigmented macules interspersed within the depigmented patch or even on normal colored skin, cafe-au-lait macules and axillary and/or inguinal freckles. In contrast to depigmentation, the pathogenesis and genetic mechanism for development of hyperpigmentation in piebaldism is ill-understood. One group has hypothesized that mutation in KIT-proto-oncogene in piebaldism leads to inadequate phosphorylation of sprouty-related, ena/vasodilator-stimulated phosphoprotein homology-1 domain containing protein 1 (SPRED1) leading to the loss of function of SPRED1. [5] This induces the development of cafe-au-lait macules and intertriginous freckles. This hypothesis remains to be proven.
There are some reports of the association of piebaldism with neurofibromatosis 1 (NF1) [6],[7],[8] or Legius syndrome. [9] In all such cases, diagnosis was based on the presence of cafe-au-lait macules and intertriginous freckles and none of the cases had any other features of NF1 such as neurofibromas or Lisch nodules and mutations in NF1 tumor suppressor gene or in SPRED1 gene. Legius syndrome is a milder phenotype of NF1 presenting with multiple cafe-au-lait macules, axillary or inguinal freckles, macrocephaly and learning difficulties but lacks many typical NF1 findings such as neurofibromas, optic gliomas and Lisch nodules. This is caused by an autosomal dominant mutation of SPRED1 gene.
Whether cases of piebaldism also presenting café-au-lait macules and intertriginous freckles represent association of piebaldism with NF1 is controversial. Proper diagnosis of associated NF1 (that carries a significant effect on health) is vital in piebaldism. There is a proposal for using stringent criteria for the diagnosis of NF1, [10] especially non-pigmentary clinical features and confirmation with mutation analysis. On the other hand, Duarte et al., state that neurofibromas may be less common in piebaldism and sensitivity of the molecular diagnosis of NF 1 mutation may not be dependable in 100% of cases [11] with the possibility of missing the diagnosis of associated NF1. Extensive freckles on limbs may mimic some reticulate pigmentary disorders e.g. Dowling Degos syndrome, dermatopathia pigmentosa reticularis and reticulate acropigmentation of Kitamura. These conditions were ruled out clinically in our patients.
In these familial cases of piebaldism, the point of particular interest was the atypical and reverse distribution of freckles that spared the classical intertriginous sites like axilla and inguinal folds but affected the limbs. This variation of an apparently banal manifestation could provide some insight into the intricate genetic control of the disease. This presentation has possibly not been reported previously. A similar pattern of distribution of freckles in both daughter and father indicated that this was not a chance finding.
In contrast to depigmentation, hyperpigmented lesions like cafe-au-lait macules and freckles are not constant features of piebaldism and their severity does not parallel the severity of the depigmentation. The present cases showed the extreme variation in the manifestation of freckles though the depigmented patches were typical of piebaldism. A separate, yet unidentified locus probably controls for the hyperpigmented macules of piebaldism, distinct from the locus that controls depigmentation. This hypothesis of separate genetic control for the depigmented and hyperpigmented components has two implications. One, it can explain variations of the phenotypic pattern of freckles independent of depigmentation. Secondly, "twin spotting" as the mechanism for development of pigmented macules within and around depigmented patches, another very classical and distinctive form of hyperpigmentation in piebaldism, appears possible. However further genetic studies are necessary to confirm this hypothesis.
| 1. |
Sánchez-Martín M, Pérez-Losada J, Rodríguez-García A, González-Sánchez B, Korf BR, Kuster W, et al. Deletion of the SLUG (SNAI2) gene results in human piebaldism. Am J Med Genet A 2003;122A: 125-32.
[Google Scholar]
|
| 2. |
Fleischman RA. Human piebald trait resulting from a dominant negative mutant allele of the c-kit membrane receptor gene. J Clin Invest 1992;89:1713-7.
[Google Scholar]
|
| 3. |
Davis BK, Verdol LD. Expansion and contraction of hypomelanotic areas in human piebaldism. Hum Genet 1976;34:163-70.
[Google Scholar]
|
| 4. |
Spritz RA, Itin PH, Gutmann DH. Piebaldism and neurofibromatosis type 1: Horses of very different colors. J Invest Dermatol 2004;122:xxxiv-xxxv.
[Google Scholar]
|
| 5. |
Chiu YE, Dugan S, Basel D, Siegel DH. Association of piebaldism, multiple café-au-lait macules, and intertriginous freckling: Clinical evidence of a common pathway between KIT and sprouty-related, ena/vasodilator-stimulated phosphoprotein homology-1 domain containing protein 1 (SPRED1). Pediatr Dermatol 2013;30:379-82.
[Google Scholar]
|
| 6. |
Duarte AF, Mota A, Baudrier T, Morais P, Santos A, Cerqueira R, et al. Piebaldism and neurofibromatosis type 1: Family report. Dermatol Online J 2010;16:11.
[Google Scholar]
|
| 7. |
Angelo C, Cianchini G, Grosso MG, Zambruno G, Cavalieri R, Paradisi M. Association of piebaldism and neurofibromatosis type 1 in a girl. Pediatr Dermatol 2001;18:490-3.
[Google Scholar]
|
| 8. |
Chang T, McGrae JD Jr, Hashimoto K. Ultrastructural study of two patients with both piebaldism and neurofibromatosis 1. Pediatr Dermatol 1993;10:224-34.
[Google Scholar]
|
| 9. |
Stevens CA, Chiang PW, Messiaen LM. Café-au-lait macules and intertriginous freckling in piebaldism: Clinical overlap with neurofibromatosis type 1 and Legius syndrome. Am J Med Genet A 2012;158A: 1195-9.
[Google Scholar]
|
| 10. |
Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol 1988;45:575-8.
[Google Scholar]
|
| 11. |
Duarte A, Mota A, Baudrier T, Morais P, Santos A, Cerqueira R, et al. Piebaldism and neurofibromatosis: State of knowledge. Dermatol Online J 2013;19:17.
[Google Scholar]
|
Fulltext Views
4,241
PDF downloads
1,068





![[Figure - 1]](#fig_ijdvl_2014_80_2_163_129404_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2014_80_2_163_129404_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2014_80_2_163_129404_f3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2014_80_2_163_129404_f4.jpg){kind=link}