Translate this page into:
Primary cutaneous angiosarcoma of the nose
Correspondence Address:
Vidya Kharkar
Department of Dermatology, Seth G. S. Medical College and K.E.M Hospital, Parel, Mumbai - 400 012, Maharastra
India
| How to cite this article: Kharkar V, Jadhav P, Thakkar V, Mahajan S, Khopkar U. Primary cutaneous angiosarcoma of the nose. Indian J Dermatol Venereol Leprol 2012;78:496-497 |
Sir,
Angiosarcoma is a rare, malignant neoplasm, composed of endothelial cells comprising less than 1% of all sarcomas, which can occur at any site; skin and superficial soft tissue being the most common locations. Cutaneous angiosarcomas primarily affect elderly persons predominantly males, with more than 50% occurring in the scalp or on face. We present a rare case of angiosarcoma, successfully treated with surgery.
A 66-year-old male presented with asymptomatic, slow growing red raised lesion over the nose since 1 year with a history of ulceration and bleeding from the lesion since a month. There was no history of any discoloration of skin prior to the development of these lesions or associated swellings in the neck. There was no history of any constitutional symptoms.
On clinical examination, a well-defined glistening erythematous ulcerated plaque was present on the nose which was soft, non-tender and easily bleeding on touch. Several non-tender, violaceous, smooth, satellite lesions were scattered around it [Figure - 1]. Nasal mucosa was completely spared. No cervical lymphadenopathy was found. Head and neck examination was unremarkable. Skin biopsy showed anastomosing vascular structures of variable diameter in the dermis, lined by plump endothelial cells with marked cellular and nuclear polymorphism, dissecting the collagen bundles [Figure - 2]a. On immunohistochemistry, tumor cells expressed CD31 and were negative for CD34 [Figure - 2]b, c, Melan A, HMB45, CD138 and S100. MIB1 marker study revealed low proliferation index around 2% - 3%, favoring a diagnosis of low grade angiosarcoma [Figure - 2]d. An MRI and CT scan of head and neck confirmed a soft tissue mass without any infiltration of underlying structures. No evidence of metastasis was found on further workup. TNM staging was T3, N0, M0. Wide excision followed by flap reconstruction was done.
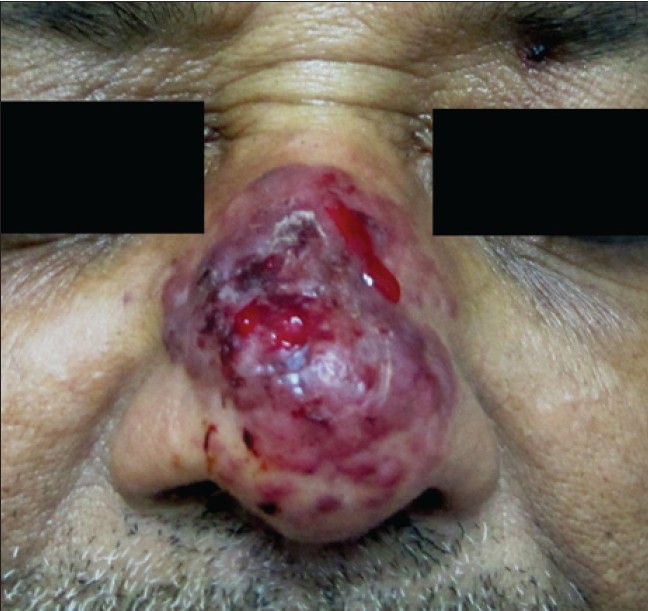 |
| Figure 1: Well-defined glistening erythematous ulcerated plaque on the nose easily bleeding on touch |
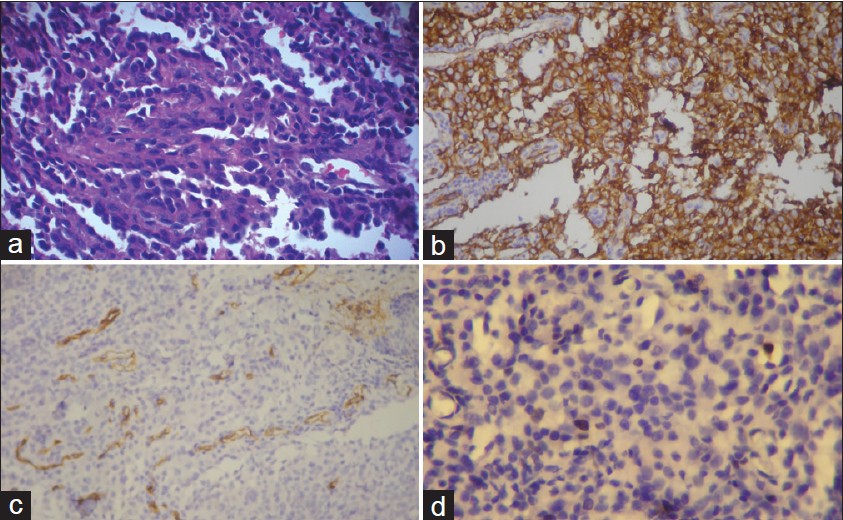 |
| Figure 2: (a) Skin biopsy showing anastomosing vascular structures in the dermis lined by plump endothelial cells with marked cellular and nuclear polymorphism dissecting the collagen bundles (H and E, ×200). (b) Immunohistochemistry studies showing tumor cells expressing CD31 (DAB Immunostain against CD31 antigen, ×200) (c) Immunohistochemistry studies showing tumor cells negative for CD34 (DAB Immunostain against CD34 antigen, ×200) (d) MIB1 marker study showing low proliferation index around 2% - 3% favoring low grade angiosarcoma (DAB Immunostain against MIB1 antigen, ×400) |
Angiosarcoma was first described in detail by Wilson Jones in 1964. Cutaneous lesions often present as ill-defined, bruise-like lesions progressing to nodular or ulcerated lesions. The most common complaint is pain or discomfort. Median size ranges from 3 to 6 cm, and over three-quarters of tumors are intermediate- or high-grade. Lung and liver are most common sites of metastases with other less common sites being lymph nodes and bone. Certain genetic alterations and complex, unbalanced karyotypes resulting in genetic losses and gains result in angiosarcomas. Gain in chromosome 5, 8, and 20 and losses of chromosomes 7, 22, and Y are commonly implicated genetic factors. [1]
They are classified as idiopathic, cutaneous AS associated with chronic lymphedema, post-radiation and miscellaneous groups. The most frequent category is the idiopathic cutaneous AS, usually seen in elderly individuals, common sites being head and neck area. They carry poor prognosis with a 5-year survival rate of 12%. [2]
Differential diagnosis of angiosarcoma includes intravascular papillary endothelial hyperplasia , kaposi sarcoma (multiple hemorrhagic sarcoma) , kaposi-like hemangioendothelioma, angiolymphoid hyperplasia: Kimura′s disease , amelanotic melanoma. [3]
Angiosarcomas are composed of irregular vascular channels, lined with atypical endothelium in a single row or several layers thick. On immunohistochemistry, these are usually positive for factor VIII-related antigen, vimentin, CD34, and CD31 tumor markers. CD31 is the most specific and sensitive marker of endothelial cell differentiation and is an important aid in diagnosis. [4]
A thorough history and physical examination should be carried out in patients suspected of having an angiosarcoma with particular attention paid to the site of origin of the primary lesion: Size, the depth of involvement, whether fixed to underlying structures, the effect on adjacent organs, nerves, blood vessels and bone. High-grade tumors should be evaluated for lung and liver metastases. Prognosis is poor. Death is due to an extensive local disease or wide spread metastasis (mainly to the lungs). Unlike other sarcomas, grade is not useful in predicting survival. No correlation exists between an appearance (e.g., ulcerated, nodular, diffuse) and survival or local recurrence. External diameter of the tumor (> 5 cm), depth of invasion (> 3 mm), mitotic rate (> 3 HPF), positive surgical margins, tumor recurrence and metastases correlate with adverse outcome. [5]
Treatment often involves an extensive surgery to achieve negative margins. Large defects are closed with skin grafts and flaps. Radical radiation therapy in the form of high-field electron beam therapy shows promise in prolonging survival of patients with localized lesions.
Doxorubicin-based chemotherapy regimens have been tried. Some major challenges in the treatment of angiosarcomas include characteristics such as, ill-defined borders, long distances of covert extension, frequency of multifocal disease and propensity for metastatic disease.
In summary, the diagnosis of angiosarcoma can be delayed due to rarity of the condition, variable presentations and puzzling tissue histology. An early detection and treatment are essential for better prognosis.
| 1. |
Schuborg C, Mertens F, Rydholm A, Brosjö O, Dictor M, Mitelman F, et al. Cytogenetic analysis of four angiosarcomas from deep and superficial soft tissue. Cancer Genet Cytogenet 1998;100:52-6.
[Google Scholar]
|
| 2. |
Morrison WH, Byers RM, Garden AS, Evans HL, Ang KK, Peters LJ. Cutaneous angiosarcoma of the head and neck. Cancer 1995;76:319-27.
[Google Scholar]
|
| 3. |
de Keizer RJ, de Wolff-Rouendaal D, Nooy MA. Angiosarcoma of the eyelid and periorbital region. Experience in Leiden with iridium192 brachytherapy and low-dose doxorubicin chemotherapy. Orbit 2008;27:5-12.
[Google Scholar]
|
| 4. |
Calonje E. Vascular tumors: Tumors and Tumor-like Conditions of Blood Vessels and Lymphatics. In: Elder DE, editor. Histopathology of the skin. 10 th ed. New Delhi: Lippincott Williams & Wilkins; 2009. p. 1007-56.
[Google Scholar]
|
| 5. |
Morgan MB, Swann M, Somach S, Eng W, Smoller B. Cutaneous angiosarcoma: a case series with prognostic correlation. J Am Acad Dermatol 2004;50:867-74.
[Google Scholar]
|
Fulltext Views
2,340
PDF downloads
781





![[Figure - 1]](#fig_ijdvl_2012_78_4_496_98086_u1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2012_78_4_496_98086_u2.jpg){kind=link}