Translate this page into:
Primary hypertrophic osteoarthropathy: Report of two novel genetic variants in the SLCO2A1 gene in two Mexican patients
2 Department of Genetics, Hospital Universitario “Dr. José Eleuterio González”, Monterrey, Nuevo León, México
3 Department of Traumatology and Orthopedics, Hospital Universitario “Dr. José Eleuterio González”, Monterrey, Nuevo León, México
Correspondence Address:
Marisol Ibarra-Ramírez
Francisco I. Madero Y Gonzalitos, Mitras Centro, Monterrey, Nuevo León 64460
México
| How to cite this article: Villarreal-Martínez A, Vázquez-Martínez OT, Martínez-de-Villarreal LE, Garay-Mendoza D, Rodríguez-Vivian C, Ocampo-Candiani J, De La Rosa-Marbán E, Ibarra-Ramírez M. Primary hypertrophic osteoarthropathy: Report of two novel genetic variants in the SLCO2A1 gene in two Mexican patients. Indian J Dermatol Venereol Leprol 2018;84:446-447 |
Sir,
Primary hypertrophic osteoarthropathy (OMIM #259100, #614441), also known as pachydermoperiostosis and Touraine–Solente–Golé syndrome, is a very rare disease characterized by the presence of pachydermia, digital clubbing and periostosis.[1]
Its pathogenesis involves genes related with prostaglandin E2 metabolism. Cases have been reported with an autosomal recessive inheritance pattern with pathogenic variants in HPGD and SCLCO2A1 genes responsible for pachydermoperiostosis type 1 and 2, respectively. In addition, some cases with an autosomal dominant pattern have been reported in which the responsible genes are unknown.[1],[2]
Here, we report two patients with novel mutations in the SLCO2A1 gene. Patient 1 was a 23-year-old male patient who attended the dermatology clinic with a 10-year history of severe acne and hyperseborrhea. On physical examination, the presence of deep furrows and thickened skin on his forehead and cheeks, acne scars, digital clubbing of hands and feet and knee swelling were noticed [Figure - 1]. An X-ray of the extremities revealed new periosteal bone formation circumferentially along the shaft of the tibia and fibula as well as cortical thickening at the distal ends.
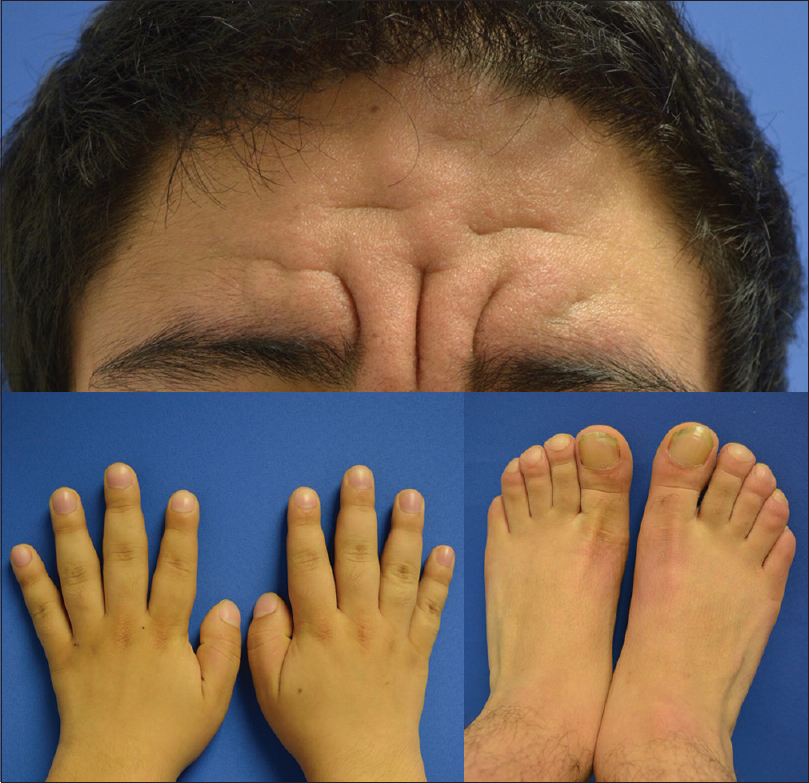 |
| Figure 1: Patient 1: Presence of deep furrows and thickened skin on the forehead and digital clubbing |
The second patient was a 24-year-old male patient with an 11-year history of bilateral knee pain, swelling and reduced range of movement. On physical examination, we noticed the presence of deep frontal furrows, thickened upper and lower eyelids, Ota nevus on the left side of the face, prognathism, bilateral knee and ankle swelling, clubbing of fingers and toes and watch-glass nails [Figure - 2]. The anteroposterior X-ray of the knee revealed a subperiosteal reaction in the lateral condyle of the femur and tibia.
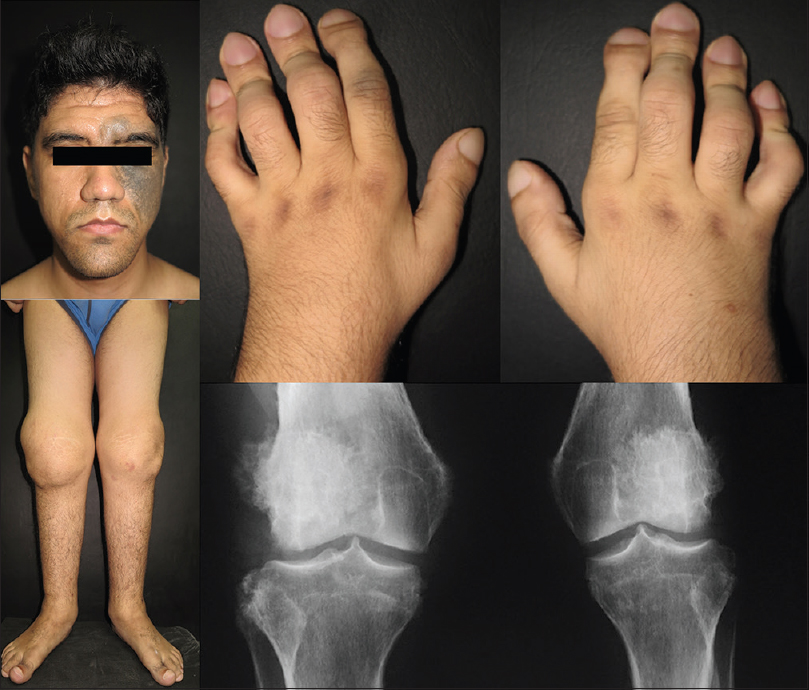 |
| Figure 2: Patient 2: Presence of deep frontal furrows, nevus of Ota on the left side of the face and prognathism, bilateral knee and ankle swelling, digital clubbing and watch-glass nails. The anteroposterior X-ray of the knee reveals a subperiosteal reaction in the lateral condyle of the femur and tibia |
Both patients lacked a family history of similar occuring. Laboratory findings, including complete blood count, thyroid function tests and serum growth hormone levels, were normal. Pachydermoperiostosis was diagnosed based on clinical findings.
Patient 1 was compound heterozygous due to a duplication of the second coding base of the SLCO2A1 gene, which was detected by Sanger sequencing and confirmed by polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP). The mutation was found in exon 1 at the start codon, and a severe effect on transcription of the gene is expected. The c.572G>C variant identified is a missense mutation that results in a change of Glycine to Alanine located at the residue position 191 where a transmembrane helix is found and where other pathogenic mutations have previously been reported.[3] The mutation is predicted to be pathogenic by PolyPhen-2 predictions. Another known polymorphism, c.1186G>A (p.A396T), has also been identified.
In the second patient, a homozygous c.96+5>A change in the first intron was identified that is close to the first exon affecting splicing, which is considered pathogenic. Mutation analysis was only performed in the father, and we found that he is a carrier of the same genetic variant.
More than 40 pathogenic variants have been described for the SLCO2A1 gene.[3],[4] Till date, no clear genotypic/phenotypic correlation exists. However, some authors have reported that in some genetic variants, such as nonsense or splicing mutations, more severe phenotypes can be observed.[5] Clinical manifestations in patients with SLCO2A1 gene mutations exhibit variable expressivity. However, it is important to highlight that most cases have been described in males and clinical characteristics appear at puberty. In patients with mutations in the HPGD gene, earlier disease initiation is noted. There are several hypotheses regarding the association of disease initiation and sex hormones, although these hypotheses have not been confirmed.[4] Although the effects of increased prostaglandin E2 on the skin are not fully understood to date, increased levels of this prostaglandin promote the development of the epidermis, epidermal hyperplasia and sebaceous gland hyperplasia in animal models.[6] In addition, a more severe phenotype is noted in patients with higher urinary prostaglandin E2 levels.[5]
In conclusion, we identified two different SLCO2A1 mutations. Further testing of mRNA expression in in-vivo models is needed to understand the effect of the genetic variants identified in these patients.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patients have given their consent for their images and other clinical information to be reported in the journal. The patients understand that name and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Giancane G, Diggle CP, Legger EG, Tekstra J, Prakken B, Brenkman AB, et al. Primary hypertrophic osteoarthropathy: An update on patient features and treatment. J Rheumatol 2015;42:2211-4.
[Google Scholar]
|
| 2. |
Niizeki H, Shiohama A, Sasaki T, Seki A, Kabashima K, Otsuka A, et al. The complete type of pachydermoperiostosis: A novel nonsense mutation p.E141* of the SLCO2A1 gene. J Dermatol Sci 2014;75:193-5.
[Google Scholar]
|
| 3. |
Lee S, Park SY, Kwon HJ, Lee CH, Kim OH, Rhee Y, et al. Identification of the mutations in the prostaglandin transporter gene, SLCO2A1 and clinical characterization in Korean patients with pachydermoperiostosis. J Korean Med Sci 2016;31:735-42.
[Google Scholar]
|
| 4. |
Hou Y, Lin Y, Qi X, Yuan L, Liao R, Pang Q, et al. Identification of mutations in the prostaglandin transporter gene SLCO2A1 and phenotypic comparison between two subtypes of primary hypertrophic osteoarthropathy (PHO): A single-center study. Bone 2018;106:96-102.
[Google Scholar]
|
| 5. |
Sasaki T, Niizeki H, Shimizu A, Shiohama A, Hirakiyama A, Okuyama T, et al. Identification of mutations in the prostaglandin transporter gene SLCO2A1 and its phenotype-genotype correlation in Japanese patients with pachydermoperiostosis. J Dermatol Sci 2012;68:36-44.
[Google Scholar]
|
| 6. |
Neufang G, Furstenberger G, Heidt M, Marks F, Müller-Decker K. Abnormal differentiation of epidermis in transgenic mice constitutively expressing cyclooxygenase-2 in skin. Proc Natl Acad Sci U S A 2001;98:7629-34.
[Google Scholar]
|
Fulltext Views
4,520
PDF downloads
1,566





![[Figure - 1]](#fig_ijdvl_2018_84_4_446_232417_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2018_84_4_446_232417_f2.jpg){kind=link}