Translate this page into:
Psoriasis in autoimmune polyendocrine syndrome type I: A possible complication or a non-endocrine minor component?
Correspondence Address:
Shital Amin Poojary
A-401, Veena Vihar, Datta Mandir Road, Dahanukar Wadi, Kandivali (West), Mumbai - 400 067, Maharashtra
India
| How to cite this article: Poojary SA, Lodha N, Gupta N. Psoriasis in autoimmune polyendocrine syndrome type I: A possible complication or a non-endocrine minor component?. Indian J Dermatol Venereol Leprol 2015;81:166-169 |
Abstract
Introduction: Autoimmune polyendocrine syndrome type I (APS I) is an autosomal recessive systemic autoimmune disorder, affecting primarily endocrine glands, in which chronic mucocutaneous candidiasis is an early and prominent manifestation. We describe the rare occurrence of unstable psoriasis (with onset of pustular lesions) in a case of APS I without mucocutaneous candidiasis. A patient presenting with unstable psoriasis (with onset of pustular lesions) was detected to have persistent hypocalcemia which led to the diagnosis of hypoparathyroidism. Subsequently he was found to have hypergonadotrophic hypogonadism, primary adrenal insufficiency (compensated), and coeliac disease, thus confirming the diagnosis of APS I. Psoriasis is very rarely reported in APS I, possibly due to the protective effect of antibodies to Th17 cytokines, which are responsible for the occurrence of candidiasis in this syndrome. However, psoriasis could occur in APS I patients without mucocutaneous candidiasis, who lack these antibodies. In our patient, possible factors aggravating psoriasis include hypocalcemia due to hypoparathyroidism as well as coeliac disease via anti-tissue transglutaminase antibodies. However, defining psoriasis as a possible minor component of APS I would require further studies of the autoimmune regulator (AIRE) gene functions.INTRODUCTION
Autoimmune polyendocrine syndrome type I (APS I; synonyms: autoimmune polyendocrinopathy candidiasis ectodermal dystrophy, Whitaker′s syndrome) is a rare autosomal recessive systemic autoimmune disorder primarily affecting the endocrine glands, which can involve several non-endocrine organs as well. [1],[2] Mucocutaneous manifestations range from the major component of early onset chronic mucocutaneous candidiasis to minor components such as vitiligo, alopecia areata, pitted nail dystrophy, enamel hypoplasia, and keratopathy. Other reported cutaneous features include urticaria-like erythema with fever, lymphoplasmacytic vasculitis, Sjogren′s syndrome and rarely psoriasis vulgaris. [1],[2],[3] Herein, we describe the rare occurrence of unstable psoriasis (with onset of pustular lesions) and its possible etiopathogenesis in a case of autoimmune polyendocrine syndrome type I.
Case Report
A 32-year-old man, born of a non-consanguineous marriage, with a history of chronic plaque psoriasis and seizure disorder, presented with multiple thick crusted plaques all over the body, associated with burning sensation and fever, of 15 days duration. Most lesions were surrounded by a zone of intense erythema and a few showed pustules along the margin [Figure - 1]a-c. Nails showed Beau′s lines, while the oral cavity was normal. A clinical diagnosis of unstable psoriasis (with onset of pustular lesions) was made and confirmed on skin biopsy [Figure - 2]. Investigations revealed anemia, leucocytosis, hyponatremia, hypochloremia, severe hypocalcemia, hyperphosphatemia, and hypoalbuminemia, all of which normalized with adequate treatment except serum calcium, which remained persistently low (<5.5 mg/dL). While in the ward, he had two episodes of generalized tonic clonic convulsions. Computed tomography (CT) scan of the brain showed large heterogenous calcifications distributed bilaterally symmetrically in the basal ganglia and caudate nucleus. Low levels of serum 25(OH) vitamin D (22 ng/mL, normal [N]:30-100) and parathormone (<3 pg/mL, N: 11-67) confirmed the diagnosis of idiopathic hypoparathyroidism as there was no past history of neck surgery, magnesium levels were normal, and there were no developmental defects. Serum calcium was brought to normal by cholecalciferol injections along with intravenous calcium gluconate. Within 2 weeks, the patient′s pustular lesions subsided and his skin lesions reverted to those of plaque psoriasis. He was started on oral acitretin, 25 mg twice a day, with which the psoriatic lesions healed completely over 3 months. During follow up, paucity of pubic and axillary hair along with grade I gynecomastia were observed. The patient, however, did not give any history of delayed puberty or erectile dysfunction. Raised levels of follicle-stimulating hormone (FSH): 20.6 mIU/mL [N: 2.5-10] and luteinizing hormone (LH): 15.6 mIU/mL [N: 2.5-10], along with azoospermia and a normal testosterone level confirmed the presence of hypergonadotropic hypogonadism. The coexistence of hypoparathyroidism and hypergonadotropic hypogonadism led us to consider the possibility of autoimmune polyendocrine syndrome type I. We screened the patient for the other common endocrine and non-endocrine components of autoimmune polyendocrine syndrome type I, although he did not have any suggestive symptoms. He was diagnosed to have compensated primary adrenal insufficiency on the basis of markedly raised adrenocorticotrophic hormone (ACTH) [313 pg/ml, N: 0-46], normal basal cortisol [13.4 μg/ml, N: 5-25], low post-ACTH cortisol [16.6 μg/ml, N > 18], the absence of adrenal mass or calcification on CT scan and normal aldosterone and renin levels. He was also diagnosed with coeliac disease based on elevated anti-tissue transglutaminase antibodies [144.02 RU/ml, N <20 RU/ml] and complete villous atrophy [Marsh grade IIIC] on duodenal biopsy. Other investigations including anti-parietal cell antibodies, intrinsic factor, anti-microsomal antibodies, blood sugars, and thyroid function tests were normal. The presence of two major components, hypoparathyroidism and Addison′s disease confirmed the diagnosis of autoimmune polyendocrine syndrome type I. Certain tests including serum interferon antibody level, 21 hydroxylase antibody level, and AIRE gene analysis could not be done due to cost and availability constraints. The patient was advised a gluten-free diet, steroid supplementation in stressful situations such as surgery, and continuation of calcium and vitamin D3.
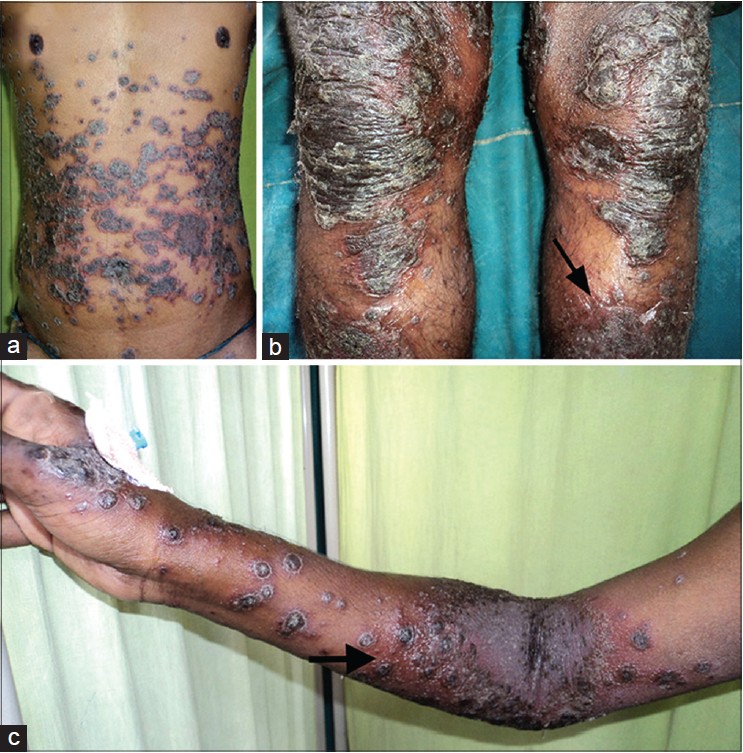 |
| Figure 1 : (a, b, c) Multiple crusted plaques, surrounded by a zone of intense erythema on chest, abdomen, both upper and lower extremities. Few pustules are seen along the border of the lesions on left leg (b) (Arrow). Maceration, erosions are seen in the cubital fossa along with pustules surrounding the plaques (c) (Arrow) |
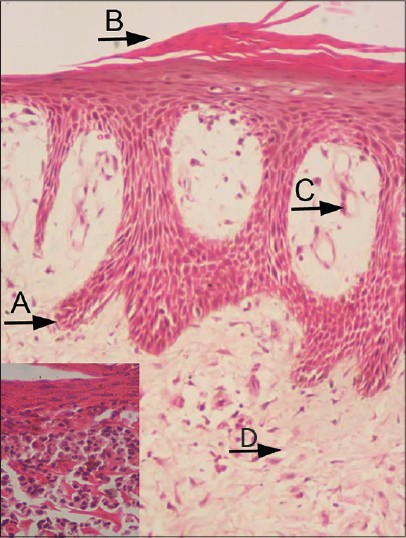 |
| Figure 2: (A) Acanthosis with elongated, test tube shaped rete ridges, (B) Parakeratosis (with a few neutrophils), (C) Suprapapillary thinning of epidermis and dilated papillary blood vessels, and (D) Superficial perivascular infiltrate (H and E, 100×). Inset: Neutrophilic microabscesses (400×) |
One year later, the patient developed an additional feature of autoimmune polyendocrine syndrome type I in the form of autoimmune thyroiditis, with raised levels of TSH [5.3 μIU/mL, N: 0.4-4] and anti-micrososmal antibodies [55.4 IU/mL, N: <34]. At that point, he was seizure free and his psoriasis was well controlled.
DISCUSSION
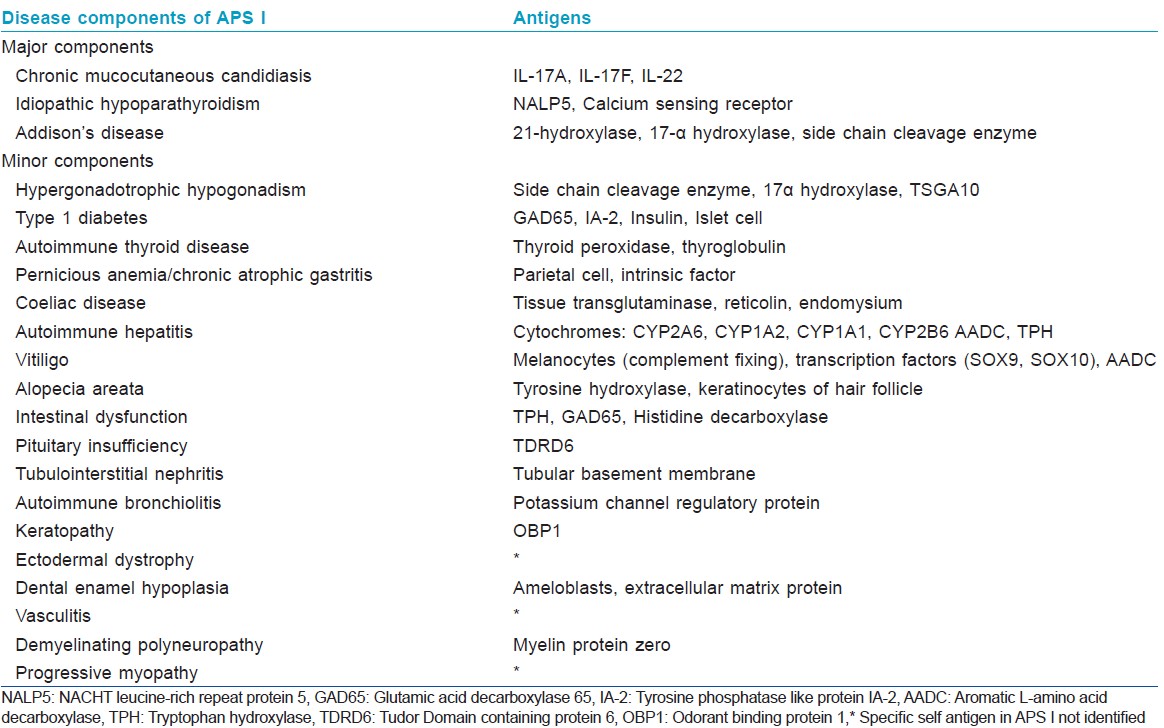
Autoimmune polyendocrine syndrome type I is caused by a mutation in the autoimmune regulator (AIRE) gene (21q22.3), which regulates thymic expression of self-antigens. This causes loss of central immunological tolerance. The resultant auto-aggressive T cells trigger inflammation in selected organs and/or production of autoantibodies against organ specific autoantigens [Table - 1] as well as consistently high titers of autoantibodies against interferonα and interferonω. [1],[2] Also, patients of autoimmune polyendocrine syndrome type I with candidiasis have severely reduced levels of interleukin-17 and interleukin-22 (Th17 response) due to neutralizing antibodies against these cytokines while those without candidiasis do not have these antibodies. [4]
In typical cases, autoimmune polyendocrine syndrome type I manifests in childhood, with mucocutaneous candidiasis being the earliest manifestation (before age 5), followed by hypoparathyroidism (by age 10) and finally adrenal insufficiency. [1],[2] Diagnostic criteria are described in [Table - 2]. [5] The chronology of the individual components is highly variable; for example, hypoparathyroidism can occur at a later age. [1]

Treatment of autoimmune polyendocrine syndrome type I is primarily focused on hormonal replacement therapy for the endocrine disturbances and oral antifungals for candidiasis. [1],[2],[5] Isolated reports have described beneficial effects of cyclosporine A, tacrolimus, methotrexate, mycophenolate mofetil, and rituximab on selected components of the syndrome. [6]
An important factor that may have played a role in the aggravation of psoriasis in our patient was persistent hypocalcemia due to hypoparathyroidism. [7] Also, the presence of coeliac disease with a high level of antitransglutaminase antibodies could have played a contributory role; tissue transglutaminase has been shown to be overexpressed in psoriatic skin and a gluten-free diet has been shown to decrease its expression by 50% with accompanying clinical improvement in psoriasis. [8]
Possible pathogenetic mechanisms linking autoimmune polyendocrine syndrome type I and psoriasis include (i) colocalization of autoimmune regulator (AIRE) with cytokeratin17 (K17). [9] K17 is a major autoantigen recognized by autoreactive T cells in psoriasis and a part of the K17/T cells/cytokine autoimmune loop [10] and (ii) deficiency of T regulatory (Treg) cell suppressor function in autoimmune polyendocrine syndrome type I. Defective Treg cell function resulting in the inability to suppress CD4 + T lymphocyte proliferative response has been implicated in pathogenesis of psoriasis. [11] However, further studies are required to confirm a direct effect of AIRE gene mutation on the immunopathogenetic pathways of psoriasis. Till then, whether psoriasis could be considered a possible non-endocrine minor component remains open to question.
The paucity of reports of psoriasis in autoimmune polyendocrine syndrome type I (although hypoparathyroidism is a common component of the syndrome) may be attributed to the protective effect of high titer antibodies to Th17 cytokines (IL-17, IL-22). Th17 cytokines play a major role in the pathogenesis of psoriasis and are also an important component of immunity against Candida. [12] Significantly, our patient did not have mucocutaneous candidiasis. This may suggest that our patient did not have antibodies to Th17 cytokines, and hence lacked their protective effect against psoriasis. This protective effect has been postulated by Kluger et al., who also suggested studies in large cohorts of autoimmune polyendocrine syndrome type I to statistically confirm the same. [13]
Thus, psoriasis as a complication or as a non-endocrine minor component of autoimmune polyendocrine syndrome type I may occur in the absence of candidiasis, that is, they may occur only as mutually exclusive phenomena.
| 1. |
Perheentupa J. Autoimmune polyendocrinopathy-candidiasis- ectodermal dystrophy. J Clin Endocrinol Metab 2006;91: 2843-50.
[Google Scholar]
|
| 2. |
Weiler FG, Dias-da-Silva, Lazaretti-Castro. Autoimmune polyendocrine syndrome type I: Case report and review of literature. Arq Bras Endocrinol Metab 2012;56:54-6.
[Google Scholar]
|
| 3. |
Cayir A, Engin RI, Turan MI, Pala E. Psoriasis vulgaris and autoimmune polyendocrine syndrome type I: A case report. J Pediatr Endocrinol Metab 2014;27:791-3.
[Google Scholar]
|
| 4. |
Kisand K, Boe Wolff AS. Podkrajsek KT, Tserel L, Link M, Kisand KV, et al. Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to TH 17 associated cytokines. J Exp Med 2010;207:299-308.
[Google Scholar]
|
| 5. |
Husebye ES, Perheentupa J, Rautemaa R, Kampe O. Clinical manifestations and management of patients with autoimmune polyendocrine syndrome type I. J Intern Med 2009;265:514-29.
[Google Scholar]
|
| 6. |
Ward L, Paquette J, Seidman E, Huot C, Alvarez F, Crock P, et al. Severe Autoimmune Polyendocrinopathy- Candidiasis-Ectodermal Dystrophy in an Adolescent girl with a novel AIRE mutation: Response to immunosuppressive therapy. J Clin Endocrinol Metab 1999;84:844-52.
[Google Scholar]
|
| 7. |
Kawamura A, Tan Kinoshita M, Suzuki H. Generalised pustular psoriasis with hypoparathyroidism. Eur J Dermatol 1999;9:574-6.
[Google Scholar]
|
| 8. |
Michaëlsson G, Ahs S, Hammarström I, Lundin IP, Hagforsen E. Gluten-Free diet in psoriasis patients with antibodies to gliadin results in decreased expression of tissue transglutaminase and fewer Ki67+cells in the dermis. Acta Derm Venereol 2003:83:425-9.
[Google Scholar]
|
| 9. |
Kumar V, Pedroza LA, Mace EM, Seeholzer S, Cotsarelis G, Condino-Neto A, et al. The AIRE which is defective in Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy patients is expressed in human epidermal and follicular keratinocytes and associates with intermediate filament protein cytokeratin 17. Am J Pathol 2010;178:983-9.
[Google Scholar]
|
| 10. |
Shi X, Jin L, Dang E, Chang T, Feng Z, Liu Y, et al. IL-17A Upregulates Keratin 17 Expression in Keratinocytes through STAT1- and STAT3-Dependent Mechanisms. J Invest Dermatol 2011;131:2401-8.
[Google Scholar]
|
| 11. |
Sugiyama H, Gyulai R, Toichi E, Garaczi E, Shimada S, Stevens SR, et al. Dysfunctional blood and target tissue CD4+CD25 high regulatory T cells in psoriasis. Mechanism underlying unrestrained pathogenic effector T cell proliferation. J Immunol 2005;174:164-73.
[Google Scholar]
|
| 12. |
Eyerich K, Foerster S, Rombold S, Seidl HP, Behrendt H, Hofmann H, et al. Patients with chronic mucocutaneous candidiasis exhibit reduced production of Th17-associated cytokines IL-17 and IL-22. J Invest Dermatol 2008;128:2640-5.
[Google Scholar]
|
| 13. |
Kluger N, Ranki A, Krohn K. APECED: Is this a model for failure of T cell and B cell tolerance? Front Immunol 2012;3:232.
[Google Scholar]
|
Fulltext Views
3,893
PDF downloads
2,314





![[Figure - 1]](#fig_ijdvl_2015_81_2_166_152285_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2015_81_2_166_152285_f2.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2015_81_2_166_152285_t3.jpg){kind=link}
![[Table - 2]](#tbl_ijdvl_2015_81_2_166_152285_t4.jpg){kind=link}