
Translate this page into:
RASopathies: Dermatologists’ viewpoints
Corresponding author: Dr. Arun C. Inamadar, Department of Dermatology, Venereology and Leprosy, Shri B. M. Patil Medical College, Hospital and Research Center, BLDE University, Vijayapur, Karnataka, India. aruninamadar@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Palit A, Inamadar AC. RASopathies: Dermatologists’ viewpoints. Indian J Dermatol Venereol Leprol 2022;88:452-63.
Abstract
Ras/mitogen-activated protein kinase pathway dysregulation results in a group of disorders, collectively termed as RASopathies. Neurofibromatosis type 1, Noonan syndrome, Noonan syndrome with multiple lentigines, Noonan syndrome/loose anagen hair, Legius syndrome, Costello syndrome, cardio-facio-cutaneous syndrome and capillary malformation-arteriovenous malformation are the well-recognized RASopathies. These are characterized by multi-organ tumours and hamartomas. Some other features in common are facial dysmorphism, skeletal abnormalities, congenital heart disease, neurocognitive abnormalities and risk of various solid-organ and haematological malignancies. Some of the RASopathies are heterogeneous, caused by several gene mutations resulting in variations in phenotypes and severity ranging from mild to fatal. Significant phenotypic overlaps among different disorders, often makes it difficult to pinpoint a clinical diagnosis. Specific cutaneous manifestations are present in some of the RASopathies and are often the earliest clinical signs/symptoms. Hence, dermatologists contribute significantly as primary care physicians by identifying disorder-specific cutaneous lesions. However, diagnostic work-up and management of these disorders are often multidisciplinary. Confirmation of diagnosis is possible only by genetic mapping in each case. Genetic counseling of the patients and the affected families is an important component of the management. The aim of this review is description of cutaneous manifestations of RASopathies in the background of multi-system involvement to enable dermatologists a comprehensive and logical approach to work up and diagnose such patients in the absence of facility for specific molecular testing.
Keywords
Capillary malformation-arteriovenous malformation syndrome
cardio-facio-cutaneous syndrome
Costello syndrome
Legius syndrome
neurofibromatosis-1
Noonan syndrome
Ras/mitogen-activated protein kinase pathway

Introduction
RASopathies include a group of disorders resulting from germline mutations of the genes regulating Ras/mitogen-activated protein kinase transduction pathway.1 These are one of the largest groups of genetic disorders brought under the term “RASopathies” (2009).2,3 Neurofibromatosis type 1 is the first RASopathy recognized.4
Some of these disorders are not so rare, prevalence being ≤1:1000.3,5 There is a wide spectrum of phenotypes and clinical overlap because of a common pathway dysregulation.3-9 Most disorders follow an autosomal dominant inheritance pattern, but other inheritance and sporadic occurrences are known.10
The understanding of the Ras/mitogen-activated protein kinase pathway function is mostly based on its role in carcinogenesis.4 Activating somatic mutations of the genes (e.g., RAS, BRAF) regulating this pathway are involved in 20–30% of human cancers.5,11-14 Such oncogenic somatic mutations are strictly confined to the neoplastic tissue or involved organ.4,11
The most commonly encountered RASopathies with unique cutaneous phenotypes are neurofibromatosis type 1, Noonan syndrome and Noonan syndrome spectrum disorders.15 Costello syndrome and cardio-facio-cutaneous syndrome are the rarest disorders.4,14 Capillary malformation-arteriovenous malformation syndrome is a recently described entity.16
Facial dysmorphism, café au lait macules, lentigines, hair anomalies, palmoplantar keratoderma, pachydermatoglaphia and vascular malformations are the usual presenting features of RASopathies. Dermatologists play an important role in recognizing the subtle distinguishing features of these overlapping phenotypic disorders and early diagnosis.
Ras/Mitogen-activated Protein Kinase Pathway
Ras are guanosine-bound small intracellular signaling proteins.4 Ras-Raf-mitogen-activated protein kinase-ERK is a highly conserved intracellular signaling pathway essential in mammalian cell development from embryogenesis through adulthood.6 This is activated by phosphorylation of ras and plays a significant role in intracellular regulation of cell differentiation, migration, proliferation, cellular aging and cell survival/death.6,11 Dysregulation of this pathway is the common pathogenic mechanism for RASopathies.6
The genes encoding the classical ras proteins are HRAS, KRAS and NRAS (rat sarcoma viral oncogene homolog).11,17,18 Germline heterogeneous mutations of various genes regulating upstream components cause mild dysregulation of this pathway resulting in most of the RASopathy syndromes [Table 1].2,4,6,11,14,15,17-24 Activating mutation of Ras and downstream effectors is to a great extent incompatible to cellular development and the resultant disorders are rare.4,15 Schematic presentation of the principal steps in the Ras-Rafmitogen-activated protein kinase-ERK signaling pathway has been given in Flow Chart 1.1,3,4,15 Apart from gene mutation, chromosomal variation may also be responsible for the genesis of RASopathies.6
| S. No. | Disorder | Gene mutation |
|---|---|---|
| 1. | Neurofibromatosis type 1 | Inactivating mutation of NF1, 17q11.2 (Neurofibromin) |
| 2. | Noonan syndrome | Activating mutation of PTPN11(50-60%),12q22 Other genes:SOS1(15%), 2p22.1; RAF1(3–17%); KRAS(<5%), 12p12;NRAS, SHOC2,10q25.2; CBL11q23.3 |
| 3. | Noonan syndrome spectrum disorders: a. Noonan syndrome with multiple lentigines b. Noonan syndrome with loose anagen hair c. Neurofibromatosis1-Noonan syndrome |
Heterozygous missense mutation of PTPN11(85-95%),12q24.12, 12q24.13;Other genes: RAF1, NRAS Missense mutation of SHOC2. p.S2G Heterozygous inactivating mutation of SPRED1,15q14 |
| 4. | Costello syndrome | Activating mutation of HRAS,11p15.5 Missense mutation at pG12S (80%) |
| 5. | Cardio-Facio-Cutaneous syndrome | Activating mutation of BRAF(75%), 7q34; MAP2K1,15q22.31and MAP2K2,19p13.3 |
| 6. | Capillary malformation-Arteriovenous malformation syndrome | Inactivating mutation of RASA1and 5q13.1-14.3 |
Rare causative genes: RIT1, SOS2, RASA2, RRAS and SYNGAP1
Newly identified rare genes: A2ML1, LZTR1, MYST4, SPRY1 and MAP3K8
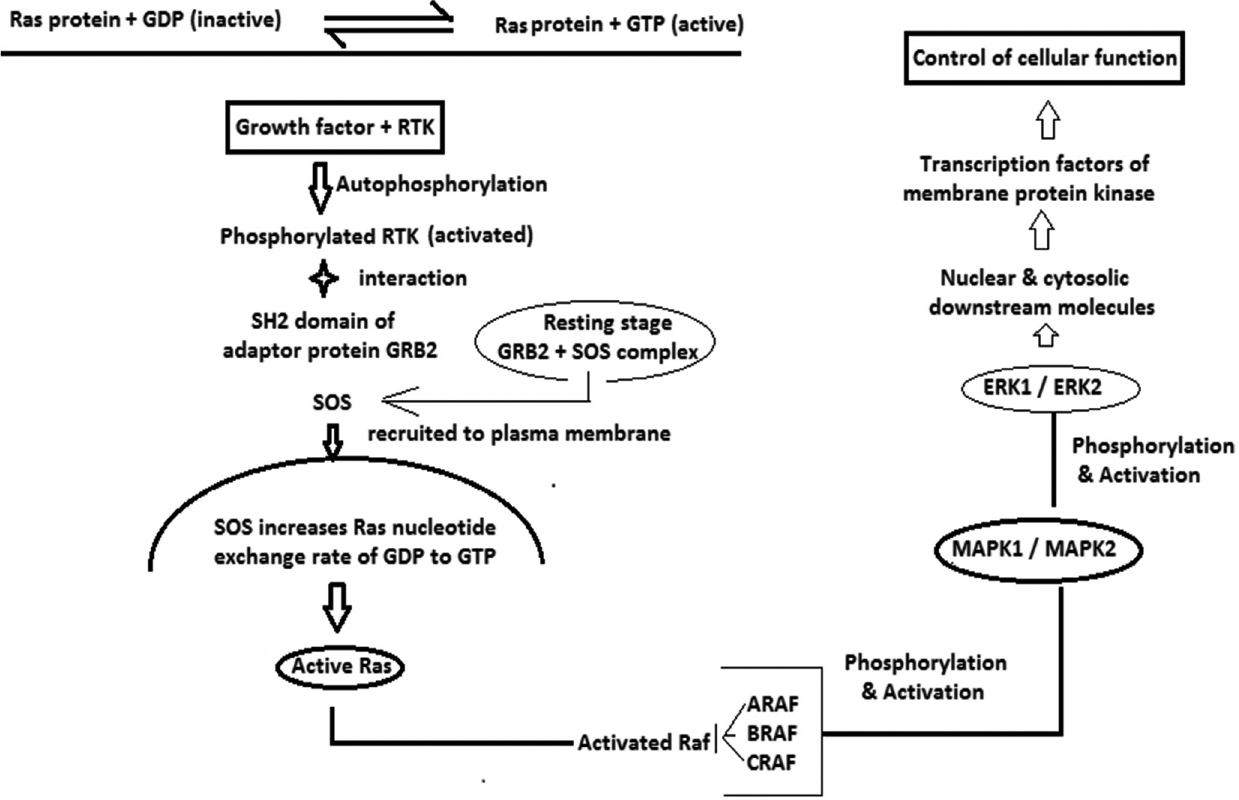
- ERK: Extracellular signal-related kinases, GDP: Guanosine di phosphate, GRB: Growth. factor receptor bound protein, GTP: Guanosine tri phosphate, MAPK: Mitogen activated protein kinase, Ras protein: Rat sarcoma protein, RTK: Receptor tyrosine kinase, ARAF, BRAF, CRAF: Raf multiprotein family; SOS: Sonof- sevenless (Guanosine nucleotide exchange factor)
Overlapping skin manifestations of Noonan syndrome, Noonan syndrome spectrum disorders and Costello syndrome have led to the proposition that this pathway dysregulation may also affect ectodermal development.2 Moreover, the distinct cutaneous features in Costello syndrome simulating acquired paraneoplastic syndromes raise the possibility of its direct pathological effect on keratinocytes.2 This pathway is also involved in learning and memory resulting in variable neuro-cognitive abnormalities in these patients.1
Mosaic RASopathies
Mosaic mutations may occur in the genes regulating Ras/mitogen-activated protein kinase pathway causing disorders distinct from those with classical germline mutation.11 These include syndromic and non-syndromic keratinocyte epidermal nevi, nevus sebaceous and Schimmelpenning syndrome.11,25,26 Mosaic mutations may occur in neurofibromatosis type 1(segmental neurofibromatosis type 1 or generalized neurofibromatosis type 1 indistinguishable from germline mutation)11,27 and Costello syndrome.28-30 The term “mosaic RASopathy” has been proposed for these conditions.11The probable explanation for mosaic RASopathies is that some germline mutations may be discordant to cell survival but may manifest in a mosaic state where limited cell numbers are affected.11,28,31
RASopathies of Dermatological Importance
Neurofibromatosis 1 (OMIM 162200)
Neurofibromatosis type 1 is the most common RASopathy (1:2500–3000 birth).4 Nearly 50% of children born with neurofibromatosis type 1 lack family history, suggestive of sporadic occurrence.32
NF1 is a very large gene encoding a ubiquitous tumour-suppressor protein neurofibromin.32 Hence, neurofibromatosis type 1 is characterized by various tumour and hamartomas developing mostly from neuro-cutaneous structures. The most important lesions include cutaneous neurofibroma and plexiform neurofibromas [Figure 1], café au lait macules [Figure 2], axillary freckling (Crowe sign), optic pathway glioma and Lisch nodules.32 Various phenotypic features, systemic and malignant associations of neurofibromatosis type 1 are presented in Tables 2 and 3.4,18,32-36
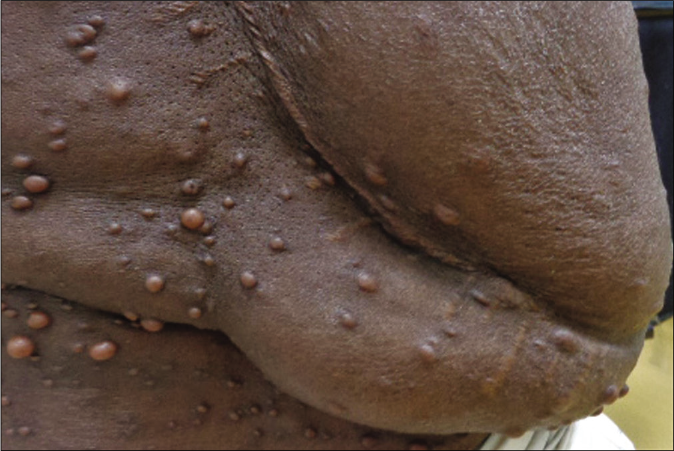
- Plexiform neurofibroma
- Light brown colour and regular borders of café au lait macules in neurofibromatosis type 1
| Involved body area/system | Phenotypic features |
|---|---|
| Structural and cranio-facial abnormalities | Short stature, Absolute macrocephaly, Hypertelorism |
| Cardiovascular | •PS (25%), Pulmonary hypertension •Cerebral and peripheral vasculopathy •Renal artery stenosis (1%) and resulting reno-vascular hypertension |
| Central nervous system | •Intracranial hamartomas, non-optic CNS astrocytoma and schwannoma •Neurocognitive abnormalities •Epilepsy |
| Ocular | •Lisch nodules (1–2 mm, yellowish brown, dome-shaped) and melanocytic hamartoma of iris (vision not impaired) |
| Oral mucosa | •NF (most commonly on tongue and grows at puberty) •Gingival overgrowth and prominent papillae of tongue (adolescents and adults) •PNF involving maxillary division of Vth cranial nerve |
| Endocrinological | Precocious puberty, Gynecomastia, Acromegaly, Addison’s disease and Hyperparathyroidism |
| Urinary system | Hydronephrosis and Double ureter |
| Skeletal | •Cortical thinning of long bones •Osteoporosis (20–40%) •Congenital long bone dysplasia (tibia), present as anterolateral bowing of leg and asymptomatic pseudoarthrosis of forearm and leg bones. Classic features •Sphenoid wing dysplasia; distinctive osteopathy of NF-1 (typically unilateral, involving orbital plate and frontal bone). Usually asymptomatic, diagnosed radiologically. May present with proptosis/pulsatile exophthalmos/temporal lobe herniation •Pectus excavatum •Scoliosis (10–28%; vertebral dysplasia) •Genu varus/valgus •Poly-/Syndactyly •Wide mandibular canal and enlarged mental foramen (radiological feature) |
NF: Neurofibroma, OPG: Optic pathway glioma, PNF: Plexiform neurofibroma, PS: Pulmonary stenosis
| Age group | Malignant solid organ tumours | Haematological malignancies |
|---|---|---|
| Childhood | •Optic pathway glioma: prevalence 5–15%; low-grade astrocytoma; commonest intracranial malignancy; mostly benign course; 33–50% develop visual symptoms •Low-grade astrocytoma; rarely glioblastoma multiforme •Rhabdomyosarcoma, neuroblastoma (7%), retinoblastoma, Wilm’s tumours |
Juvenile myelomonocytic leukemias (<5 years) |
| Adolescence | • Malignant peripheral nerve sheath tumours | |
| Adult | •Carcinoma of breast occurring in women <50 years (life-time risk 8.4%) •Malignant melanoma •Gastro-intestinal stromal tumours •Somatostatinoma •Pheochromocytoma |
Adult onset myeloid malignancies (200–500 times commoner) |
Genotype-phenotype variations
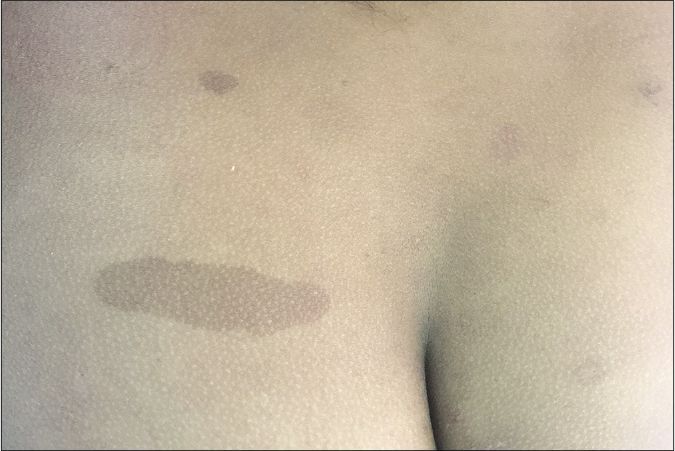
Genotype-phenotype variations in neurofibromatosis type 1 are mostly related to severity and results from deletions. A mild phenotype [Figure 3] with café au lait macules but few or without cutaneous neurofibromas, plexiform neurofibromas and optic pathway glioma occur with 3-bp microdeletion (p.Met992del,exon 17) of NF1.18,37-41 However, these phenotypes suffer from significant cognitive impairment, learning disabilities and non-optic pathway gliomas.41 A rare missense mutation of Arg1809 also present without neurofibromas, but café au lait macules and Lisch nodules are present. This phenotype is similar to Noonan syndrome with post-natal developmental delay, learning disability and pulmonary stenosis.41
- Mild phenotype of neurofibromatosis type 1 in an adult with few café au lait macules and countable cutaneous neurofibroma
Large 17q11.2 microdeletion encompassing full NF1 and the flanking genes (5–10% cases) results in the most severe phenotype with early-onset cutaneous neurofibroma, subcutaneous and spinal neurofibromas, dysmorphic face, tall for age stature with large hands and feet, hyper-flexible joints, low intelligence, muscle hypotonia, increased cardiovascular anomalies and most importantly high risk (16–26%) of malignant peripheral nerve sheath tumours18,38,42 Severe phenotype is also associated with missense mutation of codons 844–848.39,41
Diagnosis of neurofibromatosis type 1 in adolescents and adults is easier because of “tell-tale” clinical picture, but confirmation is based on available diagnostic criteria.43 All clinical features of neurofibromatosis type 1 are not manifest since birth when one or more café au lait macules are the only findings. Chronology of the appearance of important clinical features of neurofibromatosis type 1 is presented in Table 4.32,34-36,44 Some cutaneous lesions such as juvenile xanthogranuloma, nevus anemicus and glomus tumor are under consideration for inclusion in diagnostic criteria as these appear earlier.36
| Clinical feature | Approximate time of appearance |
|---|---|
| Café au lait macules | Present at birth ≥1 lesions by one year (82%); increase in size and number during first decade (≥6 lesions in infancy/childhood is an indirect evidence of future risk (95%) of developing neurofibromatosis type 1) |
| Plexiform neurofibroma | Present at birth or appear by one year of age (30%) |
| Long bone dysplasia | Present since birth; manifests by one year as bowing of the limbs, fracture and pseudoarthrosis formation |
| Optic pathway glioma | <3 years; develops within first decade |
| Freckles | Three to five years; axillae, groin, infra-mammary region and eyelids; may be generalized |
| Lisch nodules | Five to ten years; up to 2 lesions in childhood. Number of lesions increase with age, all patients have by 30 years (pathognomonic lesion) |
| Cutaneous neurofibromas | >6 years Rapid increase in the number of lesions at puberty and pregnancy |
| Kyphoscoliosis | Manifests by ten years |
| Internal neurofibroma | Adolescence; earlier in females than males |
Malignancy is the frequent cause of death and malignant peripheral nerve sheath tumors are the most common.18 These are highly aggressive spindle cell sarcomas (life-time risk 8–13%). Subtle indicators of malignant conversion in an internal neurofibroma or existing plexiform neurofibomas include unexplained pain, rapid growth, change in texture and newly developed neuro-deficit.18,36
Noonan syndrome (OMIM 163950)
Noonan syndrome is a common (1:1000–2500) widely heterogeneous disorder.3,4 In addition to autosomal dominant, autosomal recessive inheritance, sporadic occurrence and familial transmission pattern have been reported.20 Familial cases are more likely to have PTPN11 mutation.20
Genotype-phenotype variations
More than 10 various gene mutations may result in Noonan syndrome phenotype; PTPN11 is the most common followed by RAF1 and SOS1 mutations.4,17 There is significantly higher frequency of keratinization and hair disorders, generalized hyperpigmentation (adults) and pulmonary stenosis in patients with PTPN11 mutation, but minimal or no intellectual impairment17,23 RAF1 mutation is associated with macrocephaly,19 significant intellectual impairment (95%),17,19 hypertrophic cardiomyopathy,17 laryngomalacia and hoarse cry during infancy.9 Patients with SOS1 have hypertelorism and ptosis consistently,45 and mild or no cognitive impairment.17 Patients with CBL mutation are susceptible to develop myeloproliferative disorders, including juvenile myelomonocytic leukemia and those with KRAS mutations may have severe phenotype similar to Cardio-Facio-Cutaneous syndrome.17
Clinical features
The clinical picture is dominated by short stature, age-wise variable facial features and congenital heart disease.20 Post-natal growth retardation is common and the height of the affected children is less than 3rd percentile.1,20-22 In adulthood 85% of the affected individuals continue to have stunted growth.1,20,21 Various morphological features of Noonan syndrome are presented in Table 5.1,4,10,20-22,44,45
| Body area | Structural anomalies and other features |
|---|---|
| Cranium | •Relative macrocephaly (>75thpercentile); broad frontal bone •High arched palate, micrognathia, prognathism and malocclusion of teeth •Multiple dental anomalies •Craniosynostosis (sagittal variant the most common) •Multiple giant cell lesions involving soft tissues and bone of cranio-facial region |
| Neck | •Infants and children: short, webbed neck (55%) with excess nuchal skin (pterygium colli, 37%), low posterior hair-line (55%) •Adults: neck longer, webbing more prominent and prominence of trapezius muscles |
| Face (changing features with age) | At birth and early infancy: relatively unremarkable face |
| Later in infancy and childhood (facial features most prominent) •Small face underneath the large head, wide forehead and narrow temples (Tucked appearance) •High arched/diamond-shaped eyebrows •Hypertelorism (present in all patients)± telecanthus •Horizontal/down-slanting palpebral fissures (95%) with epicanthal folds; thick “hooded” eyelids, bilateral ptosis •Light-coloured/blue iris (different from parents) •Broad nasal root with bulbous tip and narrow nostrils •Upturned lips (cupid bow appearance, 95%), deeply grooved philtrum •Excessive gingival exposure on smile •Macroglossia, glossoptosis (obstructive sleep apnea) •Dysmorphic ears (90%); low set, posteriorly rotated pinnae, oval and thick helices •Relatively expression-less face |
|
| Adolescence: facial features sharper as compared to childhood •Broad upper part of face with small, pointed chin (triangular) |
|
| Adult: may become unremarkable, except a few constant features •Hypertelorism, ptosis, arched eyebrows and dysmorphic ear (static features throughout ages) •Broad forehead with recessed frontal hair-line •Deep nasolabial folds |
|
| Thorax (75%) | •Pectus deformity (typical); pectus carinatum in the upper part and pectus excavatum in the lower part. •Low-set, wide-spaced nipples (Telethelia) [Figure 4a] •Winging of the scapula |
| Spine | •Scoliosis (10–15%) •Fusion of cervical spines •Less common; kyphosis, spina bifida |
| Extremities | •Rounded shoulders •Cubitus and genu valgus •Talipes equinovarus (10–15%) •Radio-ulnar synostosis •Sausage-like digits due to peripheral lymphedema |
| Genitalia | •Uni-/bilateral cryptorchidism (80%) with impaired fertility •Hypoplastic ovary in women, with normal fertility |
Cutaneous manifestations
Patients with classic Noonan syndrome may have subtle cutaneous manifestations. Adult patients have thin, transparent and lax skin. Low posterior hair line [Figure 4b], palmo-plantar hyperkeratosis, café au lait macules, lentigines and nevi (approximately 10% of patients with KRAS mutations) are present.20-22 In a recent prospective, multicentric study among 129 confirmed Noonan syndrome patients, it was observed that multiple lentigines, > 3 café au lait macules, significantly higher keratinization and hair disorders, such as keratosis pilaris, ulerythema ophryogenes, sparse scalp hair and sparse or absent eyelashes are more consistently associated with PTPN11 mutations.22
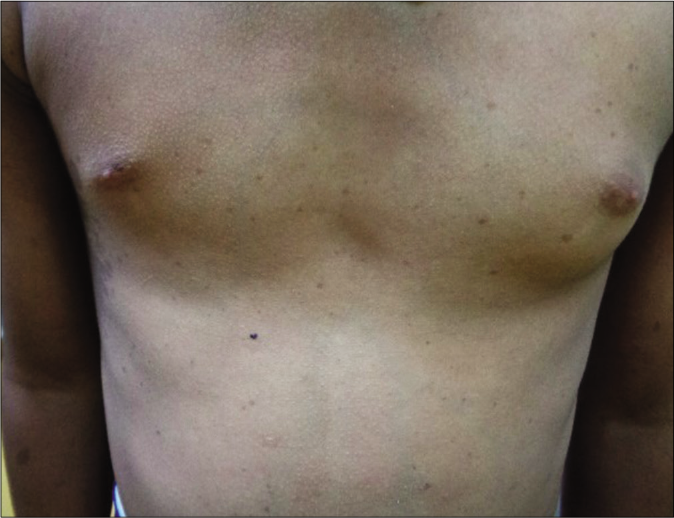
- Widely-spaced nipples in a boy with suspected Noonan syndrome
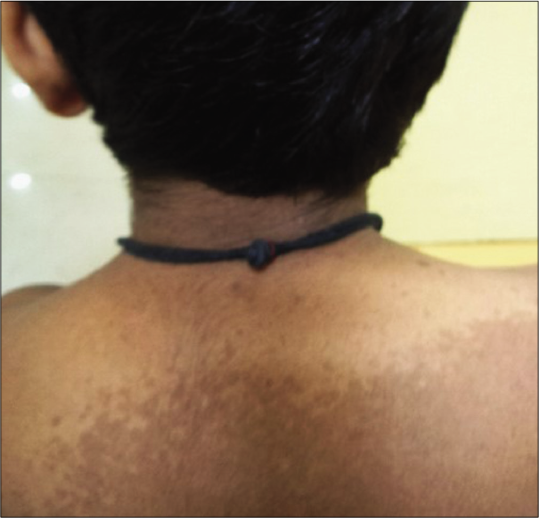
- Low posterior hair line in the same boy as in Figure 4a
Ocular manifestations
In a retrospective study among a large cohort of patients with Noonan syndrome, ocular abnormalities were frequent.45 Permanent visual impairment due to refractive errors and optic nerve anomalies were commoner with RAF1, SHOC2 and KRAS, but not seen in patients with PTPN11 mutations.45 Other visual abnormalities recorded were proptosis, strabismus, nystagmus, prominent corneal nerves and keratoconus.45
Systemic involvement
Congenital heart disease is present in more than 80% patients.20 The classical cardiac lesion is pulmonary stenosis, others being atrial septal defect, ostium secundum type and peripheral pulmonary arterial stenosis.1,20 These cardiovascular anomalies are more frequent with PTPN11 mutation.20 Feeding difficulties in childhood, reflux, vomiting, cognitive dysfunction, poor language development, difficulties in spelling and reading may be present.20,21
Haematological findings
Patients with Noonan syndrome frequently present with bleeding tendency and easy bruising.20,22 A diverse haematological abnormalities have been found (30–72%).17 Factor XI deficiency, von Willebrand disease and quantitative and/or functional disorder of platelets are most frequently described.17
Associated malignancies
Approximately 10% Noonan syndrome may have mild bone marrow abnormalities.20 Various malignancies are seen in 4% patients, with peak during childhood.11 These patients are predisposed to various leukemia such as juvenile myelomonocytic leukemia and acute myelomonocytic leukemia.1,4,11,20 Solid organ tumors described are low grade glioma, neuroblastoma and embryonal rhabdomyosarcoma.11,20
In adults clinical suspicion of Noonan syndrome may be delayed because of subtle facial appearance.1 However, as Noonan syndrome is a common cause of congenital heart disease world-wide, it should be ruled out in all such patients presented at any age.20 In doubtful cases, checking the childhood photograph of the patient and parents may be helpful.20 Clinical and laboratory evaluation for bleeding disorders (coagulation disorder profile and platelet count) must be done at diagnosis and thereafter before any surgical intervention.17
There are indicators of fetal Noonan syndrome during pregnancy. These include polyhydramnios, fetal complications such as edema, pleural effusion and hydronephrosis.20 Pre-natal diagnosis is possible in suspected cases or in the presence of positive family history. Gestational ultrasonography may be done at 12–14 weeks, 20 weeks and third trimester.20 Increased nuchal skin fold, congenital heart disease, enlarged jugular lymphatic sac and cystic hygroma are some of the clinical indicators.20 Fetal echocardiography may be done at 18–20 weeks for screening of congenital heart disease.20Genetic analysis of the collected samples (fetal blood, chorionic villi and amniotic fluid) is confirmative.20
Noonan syndrome Spectrum Disorders
There are phenotypicaly close alikes of Noonan syndrome with various genotypes designated as Noonan syndrome spectrum disorders.37
Noonan syndrome with multiple lentigines (OMIM 1511000)
The earlier nomenclature of Noonan syndrome with multiple lentigines was LEOPARD Syndrome, (Lentigines, Electrocardiographic abnormalities, Ocular hypertelorism, Pulmonary stenosis, Abnormal genitalia, Retarded growth and following Deafness) syndrome.46,47 Specific missense mutation of PTPN11 or RAF1 constitutes an allelic disorder,23 the second frequent following Noonan syndrome.47
Facial and other structural anomalies are grossly similar to Noonan syndrome.23 Hypertelorism is a consistent feature (100%) [Figure 5].47
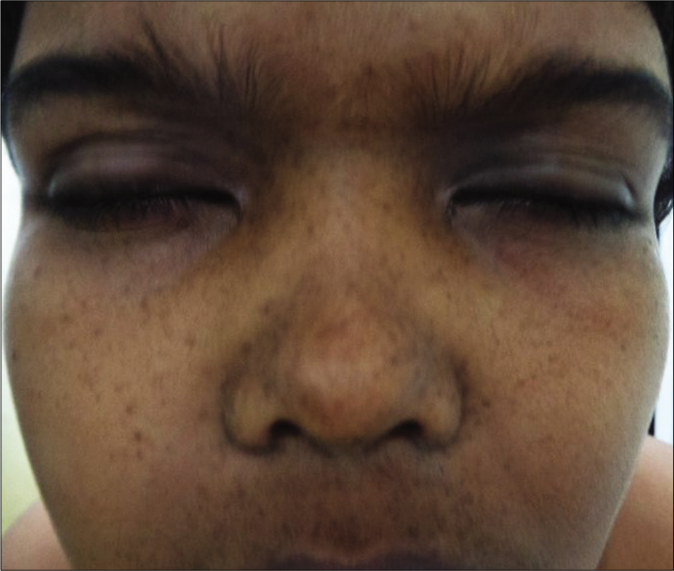
- Hypertelorism in a boy with suspected Noonan syndrome with multiple lentigines
Cutaneous features include multiple café au lait macules (>3 in 80% cases) and lentigines. café au lait macules are present since early infancy, typically darker, linear or rhomboid in shape with irregular, ragged borders [Figure 6a] in contrast to the light brown, round or oval café au lait macules of Neurofibromatosis type 1.22,23 The hallmark lentigines start appearing by five to six years of age, increase in number to more than 1000 during puberty and darken during adulthood.23 These are typically black [Figure 6b], mostly spread over the face [Figure 7] and upper trunk with accentuation on axillary and inguinal folds, palms and soles, usually sparing the mucous membranes.23 Extremities, genitalia, lips and conjunctiva may also be involved with lentigines. Café noir spots are darkly pigmented skin lesions usually ranging from 2 mm to more than 5 cm in size. Histopathologically, these may represent melanocytic nevi with or without dysplastic features or lentigo simplex.48 Nevus spilus-like lesions and multiple melanocytic nevi, more than 2 mm in diameter and more than 50 in number may be present.23
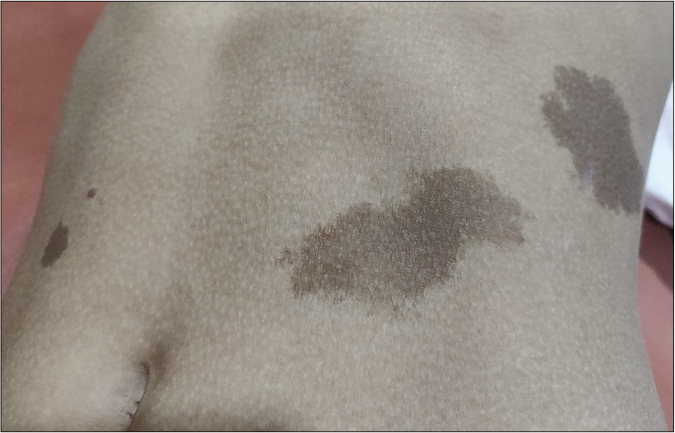
- Suspected case of Noonan syndrome with multiple lentigines, dark brown rhomboid cafe au lait macules with ragged border and cafe noir spots on the right of abdomen
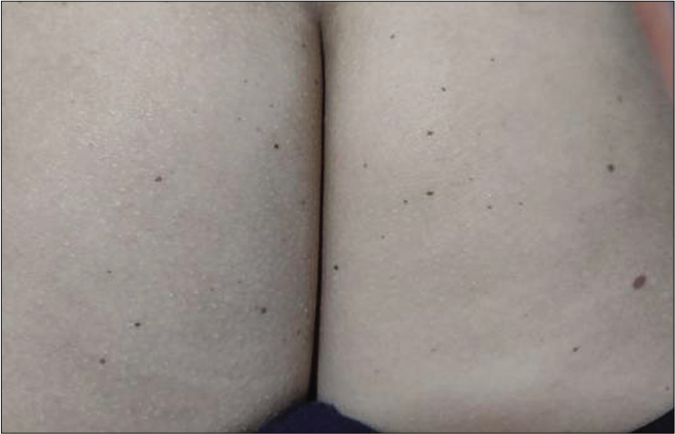
- Typical black coloured lentigines in the same patient as Figure 6a
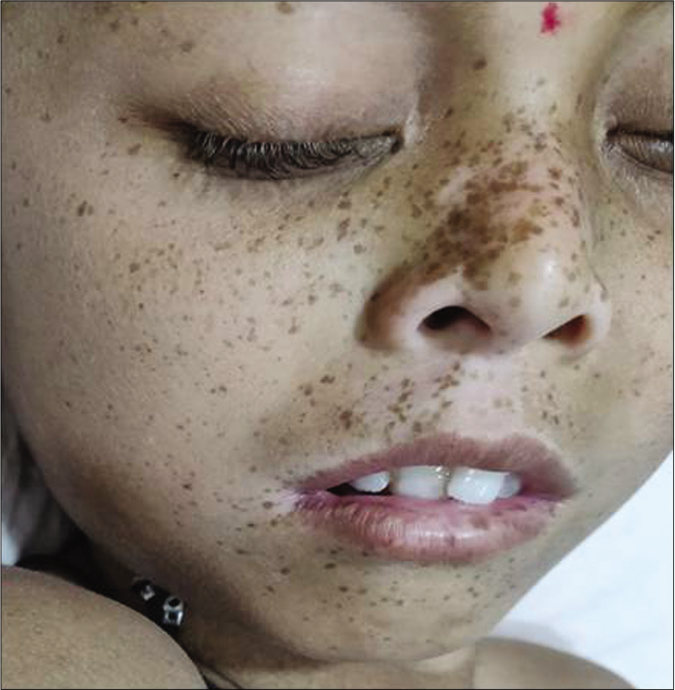
- Lentigines aggregated on face and lips with small pointed chin in a case of suspected Noonan syndrome with multiple lentigines
Left ventricular hypertrophic cardiomyopathy is more commoner (80% of the cardiac anomalies) than pulmonary stenosis in Noonan syndrome with multiple lentigines.17,23,47 Hypertrophic cardiomyopathy may be present at birth well before the appearance of skin lesions, symptoms arise over the years and may lead to life-threatening sequelae.47 Conduction abnormalities may be present in 75% cases detectable by electrocardiography.47 Sensorineural deafness may be a feature in 15–25% affected children warranting specialised care and annual evaluation.47
Noonan syndrome with multiple lentigines due to NRAS mutation, lack lentigines on face and neck; cardiac, ocular and hearing abnormalities are absent.23 Facial features are milder with macroglossia. Webbing of neck is not a common finding.44
Noonan syndrome with loose anagen hair syndrome (OMIM 607721)
Single missense mutation of SHOC2 presents with the rare phenotype Noonan syndrome with loose anagen hair.22
Frequent cutaneous findings include multiple hair anomalies such as curly or wavy, sparse and slow-growing scalp hair.22,49 The anagen follicles lack internal and external root sheath resulting in easily pluckable hair.9 However, hair morphology may alter with increasing age and may be of less diagnostic value. Temporal alopecia may be present.20,22 Thin or absent eyelashes are a frequent feature.49 Keratosis pilaris may be present.1,20 Low-birth weight (<3rd centile) and feeding difficulties are also reported.9
Legius syndrome (OMIM 601321, 611431, 609291)
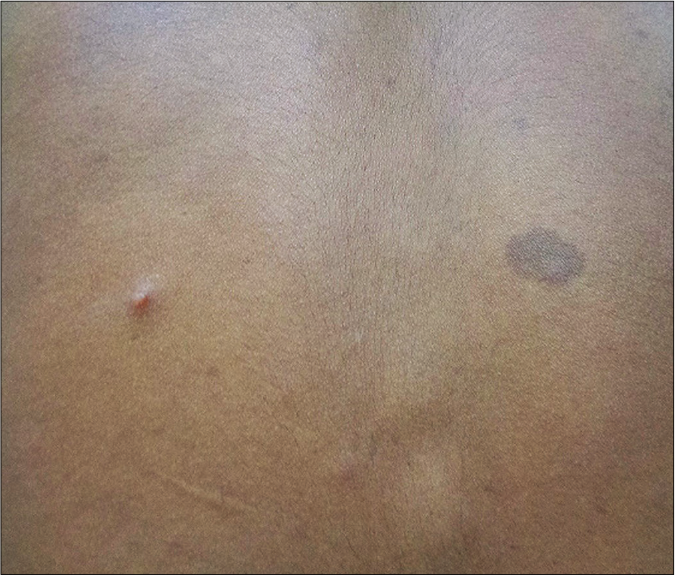
Legius syndrome has overlapping phenotypes of neurofibromatosis type 1 and Noonan syndrome. There are various mutations in SPRED1 gene and allelic variants occur.38,50 This is variably designated as neurofibromatosis type 1-like syndrome and neurofibromatosis type 1/ Noonan syndrome.37 SPRED1 is a novel regulator of NF1 function; however, inactivating mutation of SPRED1 in LS is compensated by functioning SPRED2 and SPRED3, producing a milder phenotype as compared to neurofibromatosis type 1.5 Cutaneous manifestations of Legius syndrome include multiple café au lait macules and axillary freckling [Figure 8a].4 Multiple lipomas may be present.52 However, other characteristic phenotypic features of neurofibromatosis type 1, such as cutaneous neurofibroma, plexiform neurofibomas, Lisch nodules, optic pathway glioma and malignant peripheral nerve sheath tumours are absent,1 though there is an exceptional report of optic pathway glioma in Legius syndrome.53 The facial and skeletal features are similar to Noonan syndrome; relative/absolute macrocephaly,37 hypertelorism and pectus excavatum are frequent.44,54,55 Milder learning disability, developmental delay and attention deficit hyperkinetic disorder are present.44,54,55 café au lait macules may be present in family members [Figure 8b].52 Diagnostic difficulties are common; some patients diagnosed as neurofibromatosis type 1 (2%) by fulfilling the diagnostic criteria,43 have SPRED1 mutation.55 Majority of the patients with Noonan syndrome with multiple lentigines phenotype have hypertrophic cardiomyopathy whereas cardiac abnormalities are inconsistent in Legius syndrome and do not have the higher risk of malignancies.23,36
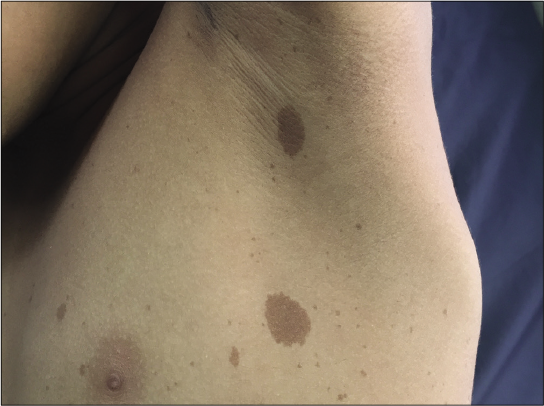
- Multiple cafe au lait macules and axillary freckling in an adolescent boy with suspected Legius syndrome.
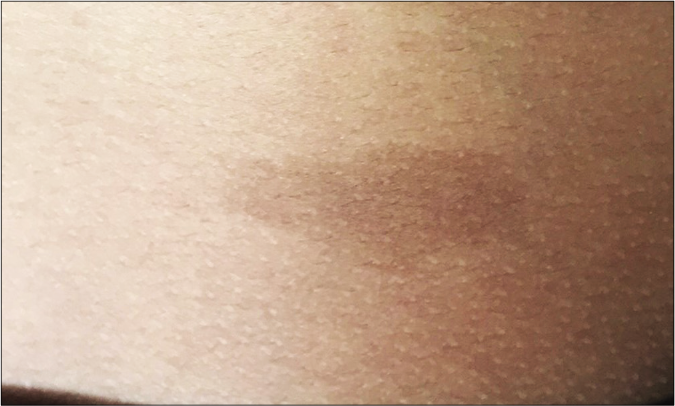
- Single cafe au lait macule in the father of the patient in Figure 8a
Costello syndrome: (OMIM 218040)
Costello syndrome is a very rare RASopathy (prevalence 1:1,290,000 birth)2,44 with unique cutaneous findings. Patients with Costello syndrome have clinical overlap with Noonan syndrome and cardio-facio-cutaneous syndrome making clinical differentiation during early life difficult. Phenotypic variations have been described in this disorder. Prenatal, severe neonatal, milder and atypical phenotypes have been described.1 Premature birth and high birth weight may be associated.4
Cutaneous findings
Cutaneous markers of Costello syndrome have been well delineated in a cross-sectional study with 46 genetically proved cases.2 Distinct morphological and cutaneous features of Costello syndrome are presented in Table 6.1,2,4,9,44,48
| Morphology | •Short stature •Relative macrocephaly •Malocclusion of jaws; open bite •Short neck with pterygium |
| Facies (distinctive) | •Coarse facies •Prominent, deeply creased and hirsute forehead •Bushy eyebrows, broad nasal tip and full cheeks •Pseudomacrostomia due to the lower face hypoplasia •Pointed chin |
| Oro-pharyngeal | •Gingival hypertrophy •Bifid uvula |
| Cutaneous papilloma | Pathognomonic skin lesion of Costello syndrome, (72%) •Appear anytime between infancy and adulthood •Warty-look (3–4 mm), multiple, verrucous pedunculated papules •Commonly around the margin of alae nasi, anterior nares and peri-anal. Eyelids, ears, neck, fingers and other moist flexures also involved •May coalesce to form hypertrophic, verrucous plaques |
| Hands and feet | •Loose redundant skin on hands and feet (characteristic feature) •Diffuse Palmoplantar keratoderma with accentuation on pressure points •Pachydermatoglyphia: deep palmo-plantar creases and stippled dermatoglyphics on distal finger pads and fingertips (honeycomb appearance) and broad spatula-like finger pads |
| Nails | •Thin with koilonychia, upturned free end •Loosely attached and fast-growing, requiring frequent clipping |
| Hair | •Scalp hair: curly/wavy, brown and brittle; very slow growth, requiring less frequent hair-cut •Tendency of frontal balding •Thick, full eyebrows due to long, unruly hairs; often requiring trimming |
| Pigmentary changes | •Darker complexion as compared to other family members; appreciable by adolescence/adulthood •Nevi and lentigines: rare; <50 in number •Acanthosis nigricans: neck and axillae (important clinical feature) |
| Sweating abnormalities(inconsistent feature) | •Excess sweating with unusual body odour (sour/vinegary) since infancy •Inadequate sweating with heat intolerance |
| Other associated cutaneous features | •Photosensitivity •Atopic eczema, (positive family history of atopy) •Earlobe creases |
Systemic features
Systemic features predominate the clinical picture in Costello syndrome leading to failure to thrive. These include severe feeding difficulty, poor suckling, reflux, hoarse voice and constipation since infancy.4,14 The affected individuals require continuous medical aid by nasogastric intubation or gastrostomy.4,14 Hypertrophic cardiomyopathy is the most common congenital heart disease. There is high frequency (10–20%) of childhood malignancies, the most common being embryonal rhabdomyosarcoma and neuroblastoma. Adolescents are prone to develop transitional cell carcinoma of urinary bladder.1,4,28 Mild to moderate intellectual disabilities are common but the affected children grow with pleasant and sociable personality beyond infancy.1,14
The combination of cutaneous papillomas, stippled pachydermatoglyphia and acanthosis nigricans in these patients are similar to that of acquired paraneoplastic syndromes.2 Cutaneous papillomas and cosmetic disfigurement due to facial lesions are often the reason for dermatology consultation during childhood.2 These are often misdiagnosed as viral warts and the underlying condition may be overlooked; re-appearance of the lesions after adequate removal is the pointer to an alternate diagnosis.
Cardio-Facio-Cutaneous syndrome: (OMIM 115150)
Cardio-Facio-Cutaneous syndrome is one of the rarest RASopathy (prevalence 1:810,000 live birth).44 Nearly all cases are sporadic, as lethality of the mutation is mostly incompatible to life and transmission.1,4 Severity of systemic involvement leads to failure to thrive.15 These include feeding difficulties, congenital heart disease (84%) and epilepsy.56 Congenital heart disease is more common compared to Noonan syndrome.56 Various associated malignancies are rhabdomyosarcoma and hepatoblastoma and also there is a higher risk for lymphoblastic leukemia and nonHodgkin’s lymphoma.11,44,56 Developmental delay, short stature, relative macrocephaly with hypoplastic supraorbital ridges and learning difficulties are similar to other RASopathies.1,9,56 Cutaneous features have been delineated in a French multicentric prospective study including 45 genetically confirmed cases of Cardio-Facio-Cutaneous syndrome.57 Scalp eczema with absence of hair is often a distinct feature at birth, followed later by growth of sparse, thin, curly or kinky hair.56,57 Eyebrows are sparse or absent.57 Painful, yellowish-orange, focal palmoplantar hyperkeratosis, diffuse keratosis pilaris, ulerythema ophryogenes, ichthyosis, multiple melanocytic nevi (appearing at puberty) and infantile hemangioma are the other cutaneous features.56,57 Typical hair morphology and mental retardation are the consistent features (100%) of cardio-facio-cutaneous syndrome.9,56 Combination of palmoplantar hyperkeratosis and ulerythema ophryogenes may be a clinical pointer toward the diagnosis of cardio-facio-cutaneous syndrome and helps in differentiation from other RASopathies with phenotypic resemblance such as Noonan syndrome and Costello syndrome.57 Pulmonary stenosis, cataract, nystagmus, hair abnormalities, ichthyosis and hemangioma are associated with BRAF mutation.9,57
Capillary malformation-arteriovenous malformation syndrome (OMIM 608354)
Capillary malformation-arteriovenous malformation syndrome is a rare disorder (1:100,000), the hallmark feature being multifocal capillary and fast-flow malformations (arteriovenous malformation syndrome and AV fistula) involving skin and other organs.1,4,16,58 Sporadic mutations are seen in 30% cases4,58 and there is variable expression in a given family.59,60 The lesions may be present at birth or develop later.61
Cutaneous findings include small and large capillary malformations.1 These are small, round or oval, pink red, punctate lesions, often surrounded by a white halo.1,60,62 Smaller lesions vary in number from 1 to 60 and are randomly distributed or localized.1,59,61 Mucosal lesions are not seen except at vermillion border of lips. Larger capillary malformations are irregular (5–22 cm), reddish brown or gray in colour.58,59 Fast-flow vascular malformations involving skin (cephalic), subcutaneous tissue, muscles, bone or viscera are present in 1/3rd cases.59,61 Deeper lesions are mostly intracranial (10%) or spinal.1,59 When fast flow lesions are present on legs, associated ParkesWeber syndrome may be considered.59 Lymphedema may be present.1
Tetralogy of Fallot, various septal and valvular defects are present in capillary malformation-arteriovenous malformation syndrome.4 Tumour susceptibility profile of capillary malformation-arteriovenous malformation syndrome is not well-delineated and may be alike to neurofibromatosis type 1.4 Complications include neurovascular accidents and congestive heart failure.3 Intracranial and spinal arteriovenous malformation syndromes manifest during childhood and associated with poor prognosis.60 Prenatal diagnosis of fast flow lesions is possible by Doppler ultrasonography.
Parkes-Weber syndrome and hereditary haemorrhagic telangiectasia may constitute close differential diagnoses.1
Conclusion
Ras/mitogen-activated protein kinase pathway disorders are getting increased attention and recognition over the past two decades. Major interest has been generated in this field due to emerging scope for targeted therapeutic interventions in recent years.1,14,39 Overlapping clinical features and phenotypic variations in the same disorder are the hurdles in early diagnosis. Age-specific appearance or subsidence of some phenotypic characters may also cause diagnostic difficulties. Some conditions are rare and phenotypic information are from small population-based studies. Diagnostic criteria are available for some of the disorders.43,59,62,63
Hallmark cutaneous manifestations are seen in some of the RASopathies. Dermatologists have a significant role in primary diagnosis or as a part of multidiscilinary team in these disorders. A clinical-feature based diagnostic approach for dermatologists is presented in Flow Chart 2. However, often multidisciplinary, investigation-based approach is essential for diagnostic work-up and management [Table 7]. Identical phenotypes resulting from different mutations in single gene or multiple genetic mutations are frequent in RASopathies. Hence, in all suspected cases of RASopathies genetic mapping is crucial for confirmation of diagnosis.38 All RASopathies are associated with great morbidity and mortality. Genetic counseling of the affected families remains a vital aspect in the management of patients with RASopathies.
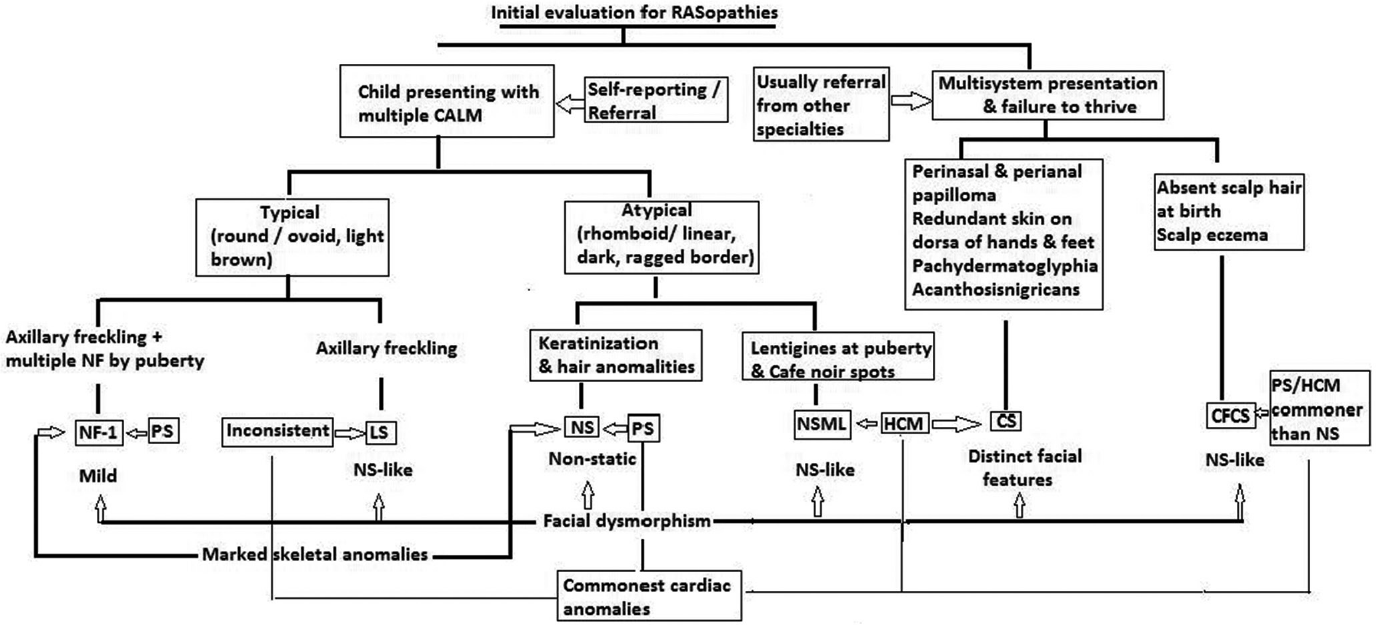
- Clinical diagnostic clues for patients presenting with overlapping features suggestive of RASopathies
- CS: Costello syndrome, CFCS: Cardio-Facio-Cutaneous syndrome, HCM: Hypertrophic cardiomyopathy, LS: Legius syndrome, NF: Neurofibroma, NF-1: Neurofibromatosis-1, NS: Noonan syndrome, NSML: Noonan syndrome with multiple lentigines, PS: Pulmonary stenosis
| RASopathy | Laboratory investigations | Imaging for systemic involvement | Multidisciplinary approach |
|---|---|---|---|
| Neurofibromatosis 1 | Complete blood count and peripheral blood smear for abnormal cells | •X-ray of long bones of forearms and legs •Organ-specific CT and MRI scan |
Neurology Ophthalmology Orthopedics Oncology |
| Noonan syndrome | Platelet count Coagulation factors Complete blood count and peripheral blood smear for abnormal cells |
•Echocardiography •Electrocardiography •X-Ray of spine and thoracic cage •Organ-specific CT and MRI scan |
Cardiology Pediatrics Hematology Ophthalmology Psychology and Behaviour therapy |
| Costello syndrome | •Echocardiography •Electrocardiography •Organ-specific CT and MRI scan |
Cardiology Gastroenterology for feeding difficulties Oncology |
|
| Cardio-Facio-Cutaneous syndrome | •Echocardiography •Electrocardiography •Organ-specific CT and MRI scan |
Cardiology Oncology |
|
| Capillary malformation-arteriovenous malformation syndrome | •Colour Doppler study in presence of large vascular malformations •Organ-specific CT and MRI scan |
Neurology and Neurosurgery Vascular surgery Cardiology for valvular/septal defects |
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Recent developments in neurofibromatosis and RASopathies: Management, diagnosis and current and future therapeutic avenues. Am J Med Genet A. 2015;167A:1-10.
- [CrossRef] [PubMed] [Google Scholar]
- Dermatological phenotype in Costello syndrome: Consequences of ras dysregulation in development. Br J Dermatol. 2012;166:601-7.
- [CrossRef] [PubMed] [Google Scholar]
- The fourth international symposium on genetic disorders of the RAS/MAPK pathway. Am J Med Genet A. 2016;178:1959-66.
- [CrossRef] [PubMed] [Google Scholar]
- RASopathies are associated with a distinct personality profile. Am J Med Genet B Neuropsychiatr Genet. 2018;174:434-46.
- [CrossRef] [PubMed] [Google Scholar]
- Expansion of the RASopathies. Curr Genet Med Rep. 2016;4:57-64.
- [CrossRef] [PubMed] [Google Scholar]
- Social skills in children with RASopathoies: A comparison of Noonan syndrome and neurofibromatosis Type-1. J Neurodev Dis. 2018;10:21.
- [CrossRef] [PubMed] [Google Scholar]
- Rasopathies: Clinical diagnosis in the first year of life. Mol Syndromol. 2010;1:282-9.
- [CrossRef] [PubMed] [Google Scholar]
- Mouse models for BRAF-induced cancers. Biochem Soc Trans. 2007;35:1329-33.
- [CrossRef] [PubMed] [Google Scholar]
- Costello and Cardio-Facio-Cutaneous syndromes: Moving toward clinical trials in RASopathies. Am J Med Genet C Semin Med Genet. 2011;157C:136-46.
- [CrossRef] [PubMed] [Google Scholar]
- Pathogenesis of the RASopathies. Hum Mol Genet. 2016;25:R123-32.
- [CrossRef] [PubMed] [Google Scholar]
- Capillary malformation-arteriovenous malformation, a new clinical and genetic disorder caused by RAS1 mutation. Am J Hum Genet. 2013;73:1240-9.
- [CrossRef] [PubMed] [Google Scholar]
- Evaluation of bleeding disorders in patients with Noonan syndrome: A systematic review. J Blood Med. 2018;9:185-92.
- [CrossRef] [PubMed] [Google Scholar]
- Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis Type 1. Lancet Oncol. 2009;10:508-15.
- [CrossRef] [Google Scholar]
- Noonan syndrome: Clinical features, diagnosis and management guidelines. Pediatrics. 2010;126:746-59.
- [CrossRef] [PubMed] [Google Scholar]
- Dermatological manifestations in Noonan syndrome: A multicentric study of 129 patients positive for mutation. Br J Dermatol. 2019;180:1438-48.
- [CrossRef] [Google Scholar]
- Grouping of Multiple-Lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am J Hum Genet. 2002;71:389-94.
- [CrossRef] [PubMed] [Google Scholar]
- RASA1: Variable phenotype with capillary and arteriovenous malformations. Curr Opin Genet Dev. 2005;15:265-9.
- [CrossRef] [PubMed] [Google Scholar]
- Mosaicism for oncogenic G12D KRAS mutation associated with epidermal nevus, polycystic kidneys and rhabdomyosarcoma. J Med Genet. 2010;47:859-62.
- [CrossRef] [PubMed] [Google Scholar]
- Postzygotic HRAS and KRAS mutations cause nevus sebaceous and schimmelpenning syndrome. Nat Genet. 2012;44:783-7.
- [CrossRef] [PubMed] [Google Scholar]
- Genetic and clinical mosaicism in a patient with neurofibromatosis Type 1. Hum Genet. 2004;114:284-90.
- [CrossRef] [PubMed] [Google Scholar]
- HRAS mutation mosaicism causing urothelial cancer and epidermal nevus. N Engl J Med. 2011;365:1940-2.
- [CrossRef] [PubMed] [Google Scholar]
- Male-to-male transmission of Costello syndrome: Inherited G12S HRAS germline mutation inherited from a father with somatic mosaicism. Am J Med Genet A. 2009;149A:315-21.
- [CrossRef] [PubMed] [Google Scholar]
- Somatic mosaicism for an HRAS mutation causes Costello syndrome. Am J Med Genet A. 2006;140A:2163-9.
- [CrossRef] [PubMed] [Google Scholar]
- Lethal genes surviving by mosaicism. A possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol. 1987;16:899-906.
- [CrossRef] [Google Scholar]
- Congenital anomalies in neurofibromatosis 1: A retrospective register-based total population study. Orphanet J Rare Dis. 2018;13:5.
- [CrossRef] [PubMed] [Google Scholar]
- Homozygous inactivation of the NF1 genes in bone marrow cells from children with neurofibromatosis Type 1 and malignant myeloid disorders. N Engl J Med. 1997;336:1713-20.
- [CrossRef] [PubMed] [Google Scholar]
- Neurofibromatosis Type 1 revisited. Pediatrics. 2009;123:124-33.
- [CrossRef] [PubMed] [Google Scholar]
- Craniofacial and oral alterations in patients with neurofibromatosis 1. Orphanet J Rare Dis. 2018;13:131.
- [CrossRef] [PubMed] [Google Scholar]
- Neurofibromatosis 1. French national guidelines based on an extensive literature review since 1966. Orphanet J Rare Dis. 2020;15:37.
- [CrossRef] [PubMed] [Google Scholar]
- Expanding the Noonan spectrum/RASopathy NGS panel: Benefits of adding NF1 and SPRED 1. Mol Genet Genomic Med. 2020;8:e1180.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical and genetic findings in children with neurofibromatosis Type 1, Legius syndrome and other related neurocutaneous disorders. Genes. 2019;10:580.
- [CrossRef] [PubMed] [Google Scholar]
- Emerging therapeutic targets for neurofibromatosis Type 1 (NF1) Expert Opin Ther Targets. 2018;22:419-37.
- [CrossRef] [PubMed] [Google Scholar]
- An absence of cutaneous neurofibromas associated with a 3-bp in frame deletion in Exon 17 of the NF1 gene (c2970-2972 del ATT): Evidence of a clinically significant NF1 genotype-phenotype correlation. Am J Hum Genet. 2007;80:140-51.
- [CrossRef] [PubMed] [Google Scholar]
- Expanding the clinical phenotype of individuals with a 3-bp in-frame deletion of the NF1 gene (c.2970_2972del): An update of genotype-phenotype correlation. Genet Med. 2019;21:867-76.
- [CrossRef] [PubMed] [Google Scholar]
- Emerging genotypephenotype relationships in patients with large NF1 deletions. Hum Genet. 2017;136:349-76.
- [CrossRef] [PubMed] [Google Scholar]
- Conference statement. National institutes of health consensus development conference. Arch Neurol. 1988;45:575-8.
- [CrossRef] [PubMed] [Google Scholar]
- A review of craniofacial and dental findings of the RASopathies. Orthod Craniofac Res. 2017;20:32-8.
- [CrossRef] [PubMed] [Google Scholar]
- Ocular findings in Noonan syndrome: A retrospective cohort study of 105 patients. Eur J Pediatr. 2018;177:1293-8.
- [CrossRef] [PubMed] [Google Scholar]
- Craniofacial and cutaneous findings in Noonan, Costello and LEOPARD syndromes. Postepy Dermatol Alergol. 2018;35:437-41.
- [CrossRef] [PubMed] [Google Scholar]
- LEOPARD syndrome: What are Café Noir spots? Pediatr Dermatol. 2008;25:444-8.
- [CrossRef] [PubMed] [Google Scholar]
- The value of dermatological phenotyping in the clinical diagnosis of RASopathies. Br J Dermatol. 2019;180:1284-301.
- [CrossRef] [PubMed] [Google Scholar]
- A shared molecular mechanism underlies the human rasopathies Legius syndrome and Neurofibromatosis-1. Genes Dev. 2012;26:1421-6.
- [CrossRef] [PubMed] [Google Scholar]
- The SPRED1 variants repository for Legius syndrome. G3 (Bethesda). 2011;1:451-6.
- [CrossRef] [PubMed] [Google Scholar]
- Legius syndrome: Case report and review of literature. Ital J Pediatr. 2015;41:8.
- [CrossRef] [PubMed] [Google Scholar]
- Case report of an optic pathway glioma in a patient with Legius syndrome. Neurooncology. 2016;18:iii78-96.
- [CrossRef] [Google Scholar]
- Observations on intelligence and behaviour in 15 patients with Legius syndrome. Am J Med Genet C Semin Med Genet. 2011;157:123-8.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical and mutational spectrum of neurofibromatosis Type 1-like syndrome. JAMA. 2009;302:2111-8.
- [CrossRef] [PubMed] [Google Scholar]
- Germline KRAS and BRAF mutations in cardio-facio-cutaneous syndrome. Nat Genet. 2006;38:294-6.
- [CrossRef] [PubMed] [Google Scholar]
- Dermatological manifestations in cardio-facio-cutaneous syndrome: A prospective multicentric study of 45 mutation-positive patients. Br J Dermatol. 2019;180:172-80.
- [CrossRef] [PubMed] [Google Scholar]
- Expanding the clinical and molecular findings in RASA1 capillary malformation-arteriovenous malformation. Eur J Hum Genet. 2018;26:1521-36.
- [CrossRef] [PubMed] [Google Scholar]
- Capillary malformation-arteriovenous malformation syndrome: Review of the literature, proposed diagnostic criteria, and recommendations for management. Pediatr Dermatol. 2013;30:409-15.
- [CrossRef] [PubMed] [Google Scholar]
- A novel RASA1 mutation causing capillary malformation-arteriovenous malformation (CM-AVM): The first genetic clinical report in East Asia. Hereditas. 2018;155:24.
- [CrossRef] [PubMed] [Google Scholar]
- CM-AVM Syndrome in a neonate: Case report and treatment with a novel flow reduction strategy. Vasc Cell. 2012;4:19.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical and molecular studies in a large Dutch family with Noonan syndrome. Am J Med Genet. 1994;53:187-91.
- [CrossRef] [PubMed] [Google Scholar]
- Multiple lentigines syndrome. Case report and review of literature. Am J Med. 1976;60:447-56.
- [CrossRef] [Google Scholar]




