Translate this page into:
Reddish-brown plaque on the left buttock
2 Skin and Stem Cell Research Center, Tehran University of Medical Sciences, Tehran, Iran
3 Department of Pathology, Jundishapur University of Medical Sciences, Ahvaz, Iran
Correspondence Address:
Maryam Aliabdi
Department of Dermatology, Azadegan Street, Imam Khomeini Hospital, Ahvaz
Iran
| How to cite this article: Yaghoobi R, Aliabdi M, Feily A, Ranjbari N, Shahriari S. Reddish-brown plaque on the left buttock. Indian J Dermatol Venereol Leprol 2012;78:231 |
A 2.5-year-old otherwise healthy boy presented to our department with an asymptomatic skin lesion originating from his left buttock since birth. The lesion had started as a small plaque 2.5 years back and later became progressive and advanced over the past 2 years. The general condition was good without any sign of growth retardation or developmental delay and there was no improvement with previous topical treatment such as steroid.
Physical examination showed a well demarcated reddish-brown plaque measuring 3.5 × 3.5 cm size on the left buttock. The lesion was firm and tender on palpation and hypertrichosis overlying its surface was noticed on close examination [Figure - 1]. Review of systems was unremarkable. Complete blood cell count with differential and platelet count were all within normal limits. A skin biopsy was obtained for the histological examination [Figure - 2] and [Figure - 3].
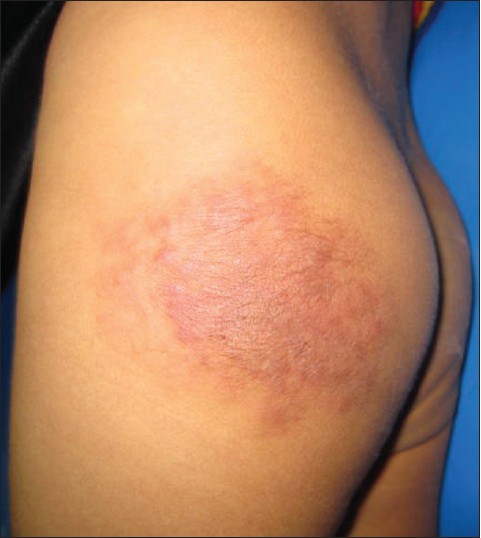 |
| Figure 1: Firm reddish-brown plaque with mottled appearance on the lateral aspect of left buttock |
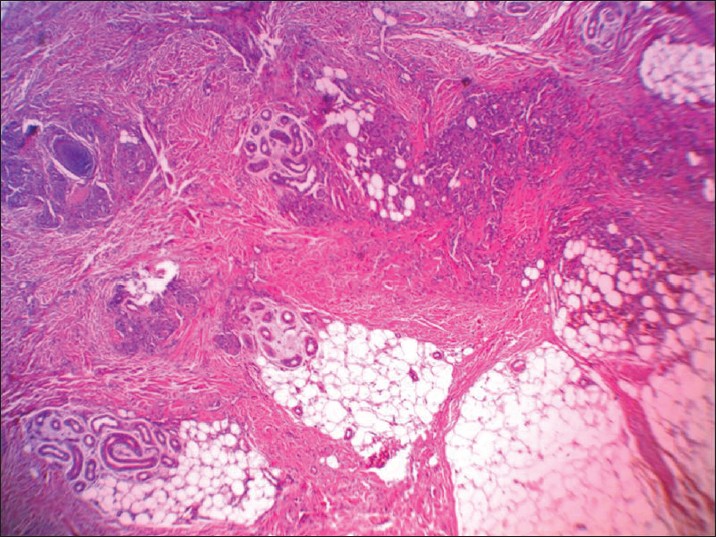 |
| Figure 2: Multiple lobules or tufts of vascular channels in dermis and subcutaneous tissue (H and E, ×20) |
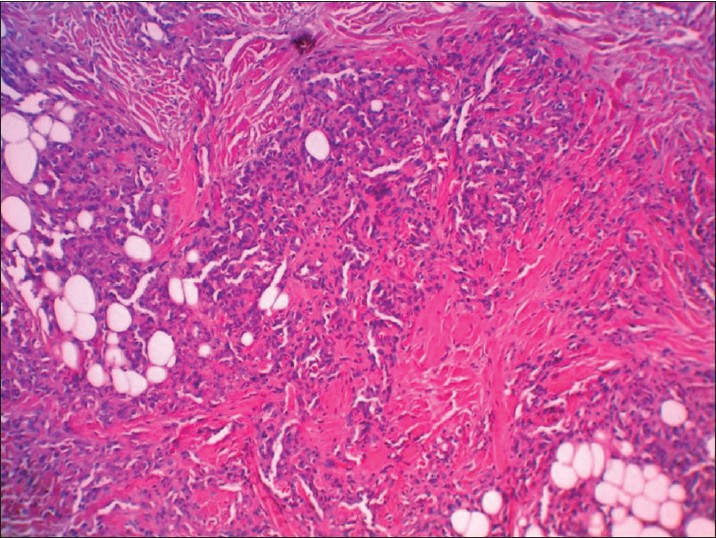 |
| Figure 3: Lobules or tufts of tumor made up of enlarged endothelial cells, without mitosis and atypicality (H and E, ×40) |
Histopathology
Histopathologic examination showed multiple separated discrete lobules within the dermis giving rise to cannonball appearance reaching the subcutis. Each of these consisted of dilated thin-walled vascular channels forming in tufts, lined by enlarged fusiform endothelial cells filled by few RBCs. Epidermis was normal looking and no inflammation or mitotic activities was seen [Figure - 2] and [Figure - 3].
What is your Diagnosis?
Answer
Diagnosis: Tufted angioma
Discussion
Tufted angioma (TA) also called angioblastoma of Nakagawa is a rare, benign vascular tumor first described in 1949. Subsequently it was described in 1971 by MacMillan and Champion as progressive capillary hemagioma. In 1976, in a report of 10 cases of similar pathology, Jones labelled them as tufted angioma. More than 200 cases have been reported, mainly in the Japanese literature and malignant transformation has never been described. About 15% of cases have a congenital occurrence and may appear at birth but about 50% occur during the 1 st year of life and a great majority appear under 10 years of age. Although in most report, TA involves both sexes equally, [1],[2],[3],[4] but in the largest series of childhood tufted angioma to date by Osio et al., [2] male sex was clearly predominant (9 of 13 patients/69%). [2] Acquired TA has also been reported in adults.
The lesions are mainly seen on the neck, upper part of the chest or shoulders, but may affect proximal portion of the limbs, face or scalp. [5],[6] TA presented with macules or plaques which enlarge progressively and then become more or less stable. Surface of the lesion may have a mottled appearance. [3] Occasionally the lesions start or persist as small violaceous papules. [6] It develops slowly and sometimes overlying hypertrichosis and hyperhidrosis are seen. [7] Tufted angiomas are usually asymptomatic but occasionally patient may have pain or tenderness on palpation. [3] Presence of tenderness, overlying skin hypertrichosis and induration can be a useful clue in differentiation of TA from ordinary hemangioma. [3] Despite its benign nature, TA can cause functional disability due to painful extensive involvement of the skin. [5]
The patients with TA may occasionally undergo complications with a serious even life threatening platelet trapping Kasabach Merritt syndrome (KMS) which is a thrombocytopenic coagulopathy due to sequestration of platelet and coagulation factors within vessels of tumor. [1],[8],[9] The frequency of KMS in TA is uncertain, but regarding the evolution of TA it may be advisable to obtain at least a complete blood cell count. Importantly, platelet count of less than 1,50,000/μ l should prompt a more extensive evaluation for coagulopathy. [2] Clinical pattern which explained chronic coagulopathy is flare of TA (infiltrating, inflammatory, and painful mass) associated with laboratory evidence of coagulopathy (defined by low fibrinogen level, high D-dimer level, and increased fibrin degeneration products) and a normal platelet count. Despite clinical improvement in the tumor, low-grade chronic consumptive coagulopathy may persist. [2]
Histopathologically, tufted angiomas are composed of lobules or tufts of tightly packed endothelial cells and capillaries with a characteristic cannonball pattern particularly in reticular dermis. There are no inflammatory cells and extension to superficial subcutis sometimes occur. [6],[10],[11] Distinguishing TA from kaposiform hemangioendothelioma (KHE) is challenging because of the morphologic and histopathologic similarities. [2] Importantly, specific marker GLUT1 in infantile hemangiomas never stains in vascular tumors such as TA. [2] Additionally, nowadays it has been shown that immunohistochemical biomarker Prox-1 is helpful in confirming the diagnosis of TA/KHE and distinguish it from infantile hemangioma and pyogenic granuloma. [12]
The other differential diagnosis include infantile hemangioma, congenital hemangioma, vascular malformations, KHE, Kaposi′s sarcoma and juvenile capillary (strawberry) angioma. [3],[7],[11]
Interestingly, TA that appear at birth or in the first year of life have a greater tendency to spontaneous regression than those that are present later in life. [7] Treatment options for TA are diverse. There is no standard therapeutic guideline for TA and choice of treatment varies from one patient to another. Eventually, because of the great tendency for spontaneous regression especially in early childhood, the best option is observation and careful follow-up to detect complications such as KMS. [2],[7] For the lesions that cause organ dysfunction or life threatening KMS or if symptoms are severe; treatment is recommended. Treatment options include: compression therapy, high dose of systemic corticosteroids, vincristine, embolization, surgical removal (choice for noncongenital small lesions), IFN α (in cases that are refractory to systemic corticosteroids), cryosurgery, pulse dye laser, chemotherapy or radiotherapy. [1],[6],[7],[10] Potent topical steroids have been used for reduction of pain. Recurrence of TA after surgery have been described. [2],[4],[9]
| 1. |
Ferrandiz-Pulido C, Mollet J, Sabado C, Ferrer B, Garcia-Patos V. Tufted angioma associated with Kasabach-Merritt phenomenon: A therapeutic challenge. Acta Derm Venereol 2010;90:535-7.
[Google Scholar]
|
| 2. |
Osio A, Fraitag S, Hadj-Rabia S, Bodemer C, Prost YD, Hamel-Teillac D. Clinical spectrum of tufted angiomas in childhood: A report of 13 cases and a review of the literature. Arch Dermatol 2010;146:758-63.
[Google Scholar]
|
| 3. |
AL-Zaabi AM , Ghazarian D, Greenberg GR, Shaw JC. Eruptive tufted angiomas in a patient with Crohn's disease. J Clin Pathol 2005;58:214-6.
[Google Scholar]
|
| 4. |
Samra N, Das S, Roy AK. Annular tufted angioma. Indian J Dermatol Venereol Leprol 2007;73:435-6.
[Google Scholar]
|
| 5. |
Silva RS, Bressan AL, Nascimento LB, Kac BK, Azulay-Abulafia L. Tufted angioma and myofascial pain syndrome. An Bras Dermatol 2011;86:125-7.
[Google Scholar]
|
| 6. |
Daley T. Acquired tufted angioma of the lower lip mucosa. J Can Dent Assoc 2000;66:137.
[Google Scholar]
|
| 7. |
Alberola FT, Betlloch I, Montero LC, Nortes IB, Martínez NL, Paz AM. Congenital tufted angioma: Case report and review of the literature. Dermatol Online J 2010;16:2.
[Google Scholar]
|
| 8. |
Reddy IS, Anuradha SV, Swarnalata G. Congenital giant tufted angioma. Indian J Dermatol Venereol Leprol 2009;75:639.
[Google Scholar]
|
| 9. |
Ateyya N, F Botros M, Abdo I, Foda S, Amer H, Saad S.Tufted angioma. Egypt Dermatol Online J 2005;1;9.
[Google Scholar]
|
| 10. |
Schaffer J, Fangman W, Bossenbroek N, Meehan AS, Kamino H. Tufted angioma. Dermatol Online J 2008;14;2.
[Google Scholar]
|
| 11. |
Wilmer A, Kaatz M, Bocker T, Wollina U. Tufted angioma. Eur J Dermatol 1999;9:51-3.
[Google Scholar]
|
| 12. |
Le Huu AR, Jokinen CH, Rubin BP, Mihm MC, Weiss SW, North PE, et al. Expression of prox1, lymphatic endothelial nuclear transcription factor, in Kaposiform hemangio endothelioma and tufted angioma. Am J Surg Pathol 2010;34:1563-73.
[Google Scholar]
|
Fulltext Views
3,615
PDF downloads
2,414





![[Figure - 1]](#fig_ijdvl_2012_78_2_231_93667_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2012_78_2_231_93667_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2012_78_2_231_93667_f3.jpg){kind=link}