
Translate this page into:
Reticulate sclerodermoid pigmentation in Hutchinson–Gilford progeria syndrome – A report of two cases
 , Biswanath Behera1, Pavithra Ayyanar3, Keshavamurthy Vinay2, Vishal Thakur1,
, Biswanath Behera1, Pavithra Ayyanar3, Keshavamurthy Vinay2, Vishal Thakur1,
Corresponding author: Dr. Vishal Thakur, Department of Dermatology and Venereology, All India Institute of Medical Sciences, Bhubaneswar, Odisha, India. drvishal87igmc@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Gowda SK, Baskaran N, Behera B, Ayyanar P, Vinay K, Thakur V. Reticulate sclerodermoid pigmentation in Hutchinson-Gilford progeria syndrome – A report of two cases. Indian J Dermatol Venereol Leprol. 2025;91:250-2. doi: 10.25259/IJDVL_1325_2023
Dear Editor,
Hutchinson–Gilford Progeria syndrome (HGPS, OMIM 176670) is a rare (prevalence 1 in 4 million births) autosomal dominant genetic premature ageing disorder due to mutation in the lamin A (LMNA) gene.1 Only about 150 cases of this disease have been reported to date. It has characteristic facial features such as a disproportionately larger head than face, delayed dentition, loss of scalp hair, delayed sexual maturity and cardiovascular abnormalities.2 Herein, we present two cases of HGPS having the unique feature of reticulate hyperpigmentation on the trunk with sclerodermoid skin changes and fat outpouching.
Our index case was a 14-month-old boy born of a non-consanguineous marriage who presented with loss of hair on the scalp and pigmentation on the abdomen, back and lower limbs since 1 month of age. There was no history of similar features among siblings or first-degree relatives. On examination, the height (73cm) and weight (7.5 kg) of the child were between the 3rd and 50th centile for his age. The face showed frontal bossing, prominent scalp veins, a sculptured nose, midfacial hypoplasia, micrognathia, protruded eyes and ears with a thin helix and wide concha [Figure 1a]. Interestingly, the child had in addition, reticulate pigmentation on the abdomen, back and lower limbs; along with binding down of abdominal skin with pebble-like outpouchings from the abdominal wall [Figure 1b]. Biopsy from the area having reticulate hyperpigmentation and outpouching of skin revealed epidermal acanthosis, increased basal layer melanin and melanophages in the superficial dermis with diffuse dermal sclerosis [Figure 2]. Whole exome sequencing confirmed the clinical suspicion of HGPS with heterozygous variation in Exon 11 of the LMNA gene in the form of single base substitution at 1824 nucleotide sequence (c.1824C>T).
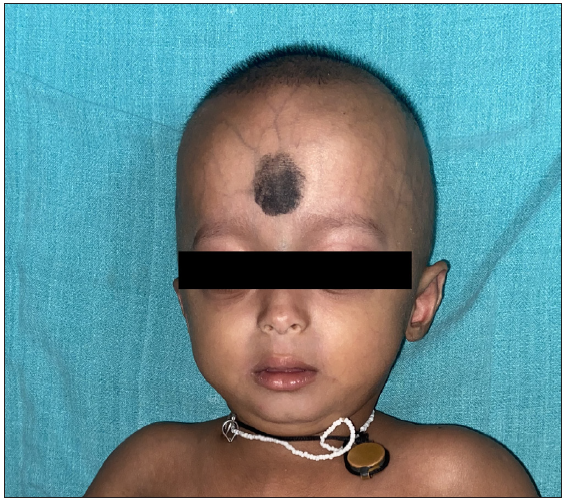
- Case 1 showing characteristic facial features including frontal bossing, sparse eyebrows and eyelashes, flat and broad nasal bridge, prominent philtrum, micrognathia and circumoral cyanosis, sparse and thin hair and prominent dilated veins visible through the translucent skin.

- Sclerodermoid changes in reticulate pattern along with mottled hyperpigmentation on the abdomen. Pebble-like outpouchings of fat on the abdominal wall (black arrows).
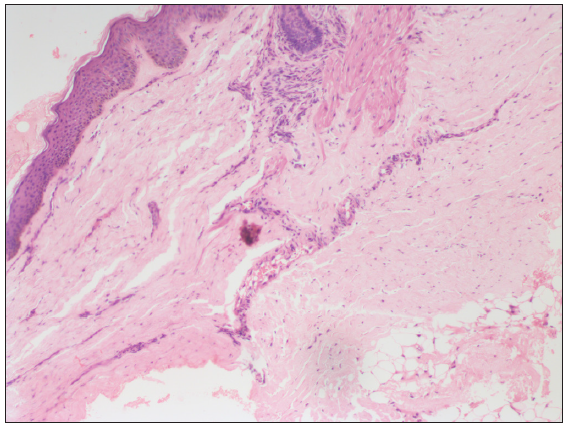
- Epidermal acanthosis, increased basal layer pigmentation and melanophages in the superficial dermis with diffuse dermal sclerosis (Haematoxylin & eosin, 100x).
Another nine-month-old girl child presented with similar cutaneous findings to our index case [Figure 3]. Whole-exome sequencing revealed a pathogenic mutation in the LMNA gene at Exon 11 variant c. 1822G> A. The rest of the systemic examinations, including ocular, hearing and cardiac examination were normal in both children.
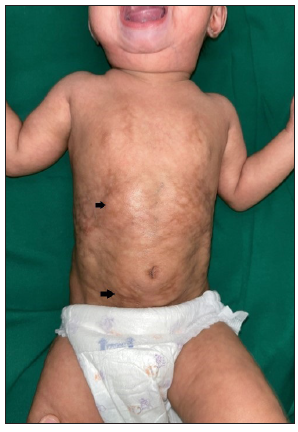
- Case 2 showing sclerodermoid skin changes, mottled hyperpigmentation and fat outpouchings (black arrows) on the abdomen.
HGPS is caused by abnormal lamin-A precursor progerin with retained farnesyl group leading to genomic instability and premature cell senescence.2 It is characterised by alopecia (100%), sclerodermoid skin changes (78%), prominent skin vasculature visible through translucent skin (22%), dyspigmentation (16%) and characteristic facies (excessive folding on forehead and cheeks, small ear lobes, protrusion of eyes, thin nasal bridge and collapsed flattened nasal tip).3 Our cases had reticulate pigmentation along with sclerodermoid skin changes prominent on the abdomen and proximal extremities.
Sclerodermoid changes in progeria initially (6 to 12 months) start as swollen and thick skin over the abdomen and proximal limbs which later (18 months) become sclerodermatous and bound down. As the disease progresses (around 2 years), the binding down vanishes and the skin becomes thin, atrophic and dry, with/without scaling.1 In a case series, the bilateral nipples and labium majus were spared during the initial sclerotic process, leading to a ‘protuberant soft nipples and labium majus’ which was labelled as ‘protuberant bikini sign’ (88.9%).4 However, in our cases, we noticed characteristic reticulate sclerodermoid hyperpigmented skin changes with outpouching of fat in uninvolved sclerotic areas giving a ‘plastic bubble wrap’ appearance.
Histopathology of sclerodermatous skin during the acute phase reveals normal epidermis, hypertrophic collagen and decreased fibroblast with scattered lymphocytes that invade the subcutaneous tissue. At a later stage, biopsy reveals thickened dermis and homogenised collagen in the deeper dermis with decreased cellularity. At the end stage, the biopsy shows replacement of the corium by fibrotic hyaline tissue, decreased sweat glands and sebaceous glands and atrophic subcutaneous adipose tissue.2
It is a fatal disease with most patients not surviving beyond 13 years due to death from stroke or myocardial infarction because of severe atherosclerosis. Management is based on counselling the parents regarding the disease prognosis and the need for regular follow-up to detect cardiovascular and skeletal abnormalities. Low-dose aspirin has been advocated as prophylaxis to delay atherosclerotic changes. FDA has recently approved lonafarnib (farnesyl transferase inhibitor) for this disorder offering hope in the management of this deadly disease.5 The drug however is not yet commercially available across many countries.
We report these cases to highlight the under-reported but characteristic cutaneous finding of reticulate sclerodermoid pigmentation and fat outpouchings in patients of HGPS. This morphological finding can help as a cutaneous marker of HGPS in resource-limited settings where facilities for genetic diagnosis are not easily available.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Hutchinson-Gilford Progeria Syndrome: A premature aging disease. Mol Neurobiol. 2018;55:4417-27.
- [CrossRef] [PubMed] [Google Scholar]
- Hutchinson-Gilford progeria syndrome: Review of the phenotype. Am J Med Genet A. 2006;140:2603-24.
- [CrossRef] [PubMed] [Google Scholar]
- Initial cutaneous manifestations of Hutchinson-Gilford progeria syndrome. Pediatr Dermatol. 2014;31:196-202.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Clinical and genetic features of children with Hutchinson-Gilford progeria syndrome: A case series and a literature review. J Eur Acad Dermatol Venereol. 2021;35:e387-e91.
- [CrossRef] [PubMed] [Google Scholar]
- Lonafarnib: First approval. Drugs. 2021;81:283-9.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




