
Translate this page into:
Solitary purpuric plaque in a four-year-old girl: Histopathological diagnostic challenge
Corresponding author: Associate Professor, Pinar Incel Uysal, Department of Dermatology, TOBB ETU Faculty of Medicine, Yasam cad. No. 5, Ankara 06560, Turkey. pinarincel@hotmail.com
-
Received: ,
Accepted: ,
How to cite this article: Uysal PI, Ayva ES, Tepeoglu M, Uysal AC. Solitary purpuric plaque in a four-year-old girl: Histopathological diagnostic challenge. Indian J Dermatol Venereol Leprol 2022;88:541-3.
Sir,
Lichen aureus with lymphocyte atypia and monoclonal rearrangement is considered as a mimicker of mycosis fungoides. Both disorders; lichen aureus and mycosis fungoides, are rarely recognized in childhood. Here, we describe a healthy girl with a progressive red-brown plaque posing a histopathological challenge.
A four-year-old girl presented with three-week history of asymptomatic eruption on her left axilla. She was otherwise healthy and on no systemic medication. She was participating in a gymnastic class for a few months. The patient had no previous history of infection or exposure to contact allergen. Dermatological examination revealed a solitary reddish plaque with sharp demarcation on her left axilla [Figure 1]. There was no lymphadenopathy or hepatosplenomegaly. Laboratory parameters were within normal limits.
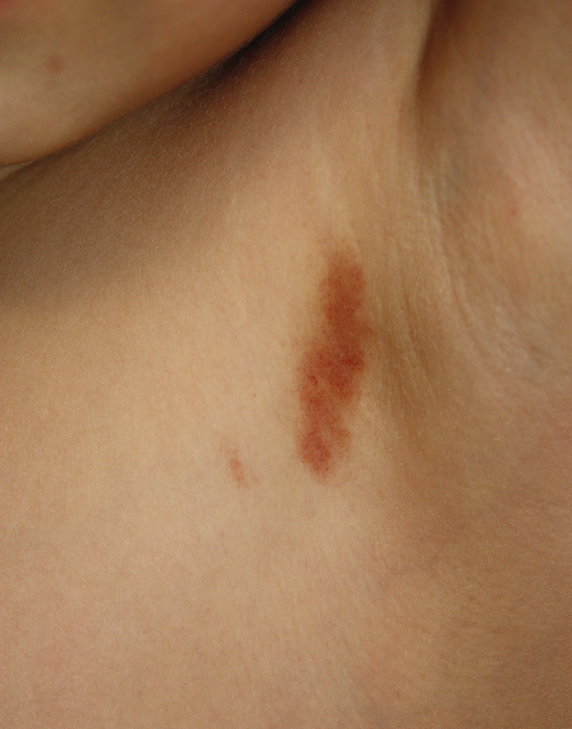
- The plaque in left axilla
Histopathological evaluation of the plaque revealed a moderately dense upper dermal lymphoid infiltrate with intraepidermal lymphocytes tagging the dermo-epidermal junction [Figures 2a and b]. A few intraepidermal lymphocytes were observed. The lymphocytes showed variable nuclear pleomorphism, nuclear membrane irregularity and hyperchromasia with haloes around the nuclei. There were a few clusters of atypical lymphocytes with minimal spongiosis [Figure 2c]. Immunohistochemically, the lymphocytes expressed pan-T-cell markers, CD2, CD3, CD5 and CD7. A substantial reduction in CD7 expression was also observed [Figures 3a and b]. CD8+ phenotype was more prominent than CD4+ phenotype in the lymphoid population [Figures 3c and d]. Monoclonality was detected in the T-cell receptor gene rearrangement study. She was also consulted by a paediatric haematologist. Further detailed clinical and radiological evaluation revealed no abnormality. The patient was treated with local corticosteroids with substantial improvement.
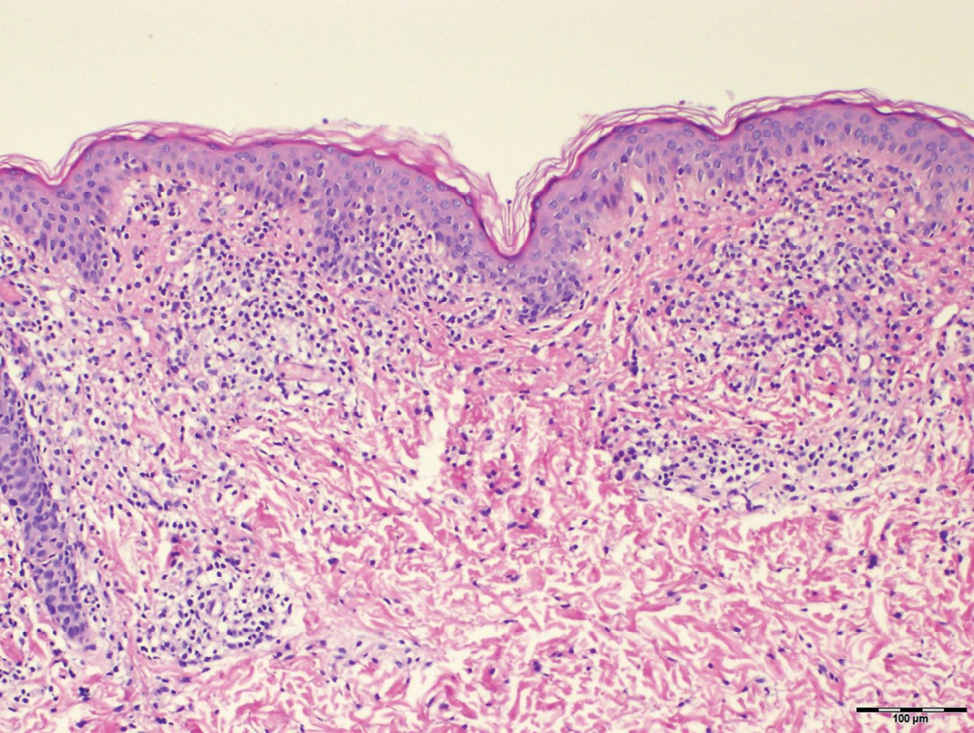
- Band-like dermal lymphoid infiltration (hematoxylin and eosin, ×40)
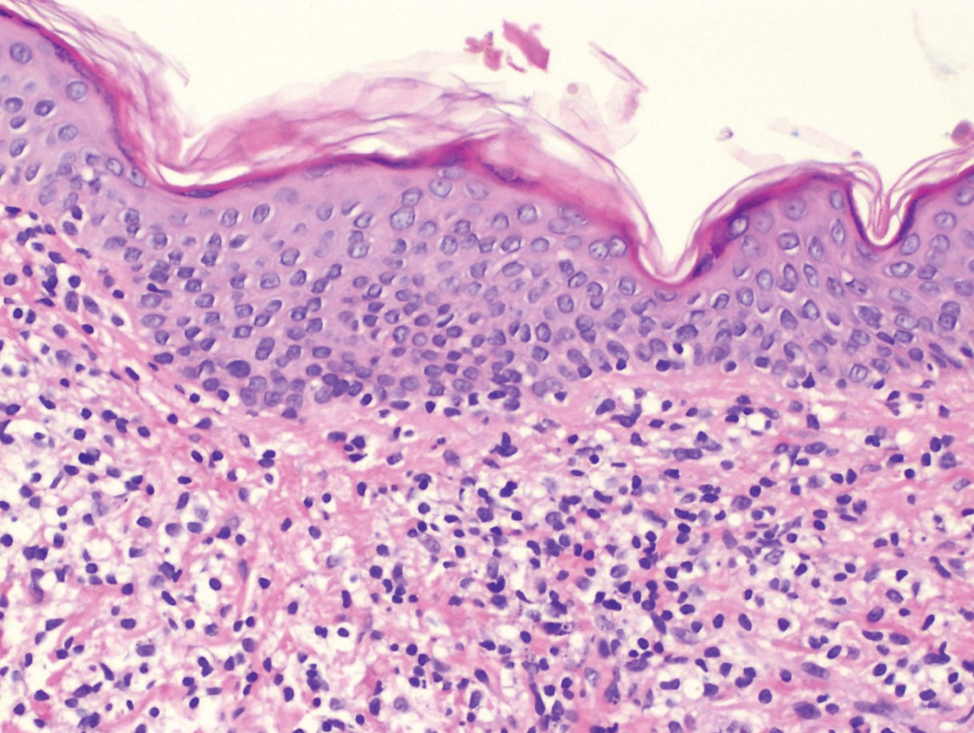
- Intraepidermal lymphocytes tagging at the dermo-epidermal junction (hematoxylin and eosin, ×100)
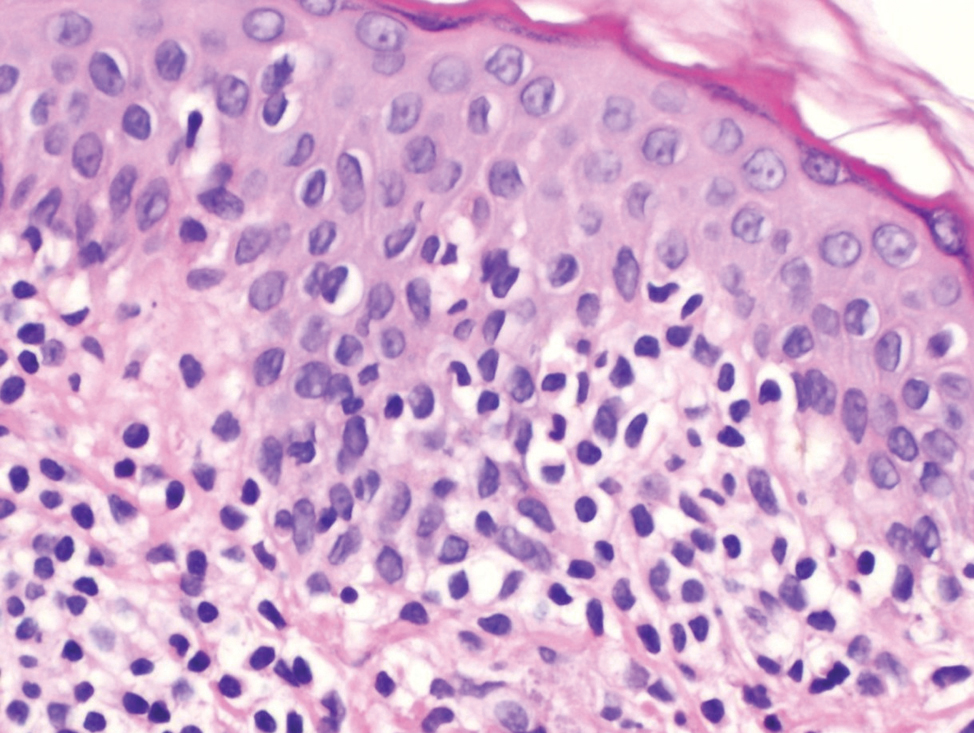
- Intraepidermal lymphocytes showing variable nuclear pleomorphism with perinuclear haloes (hematoxylin and eosin, ×200)
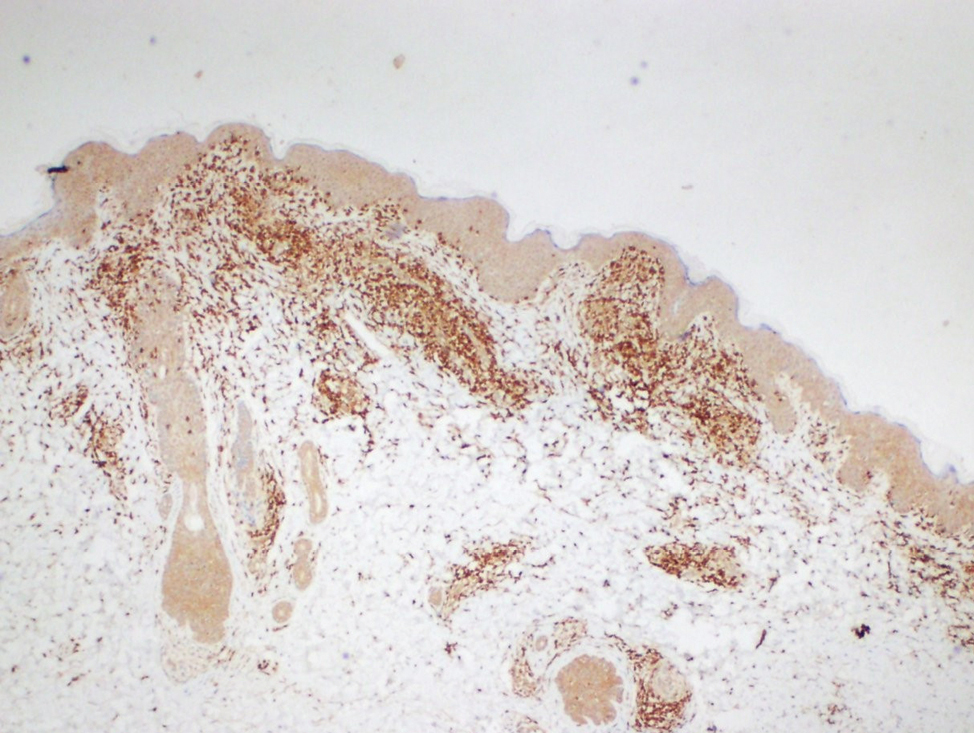
- The lymphocytes immunohistochemically express CD3 which is pan T marker (Diaminobenzidine, ×40)
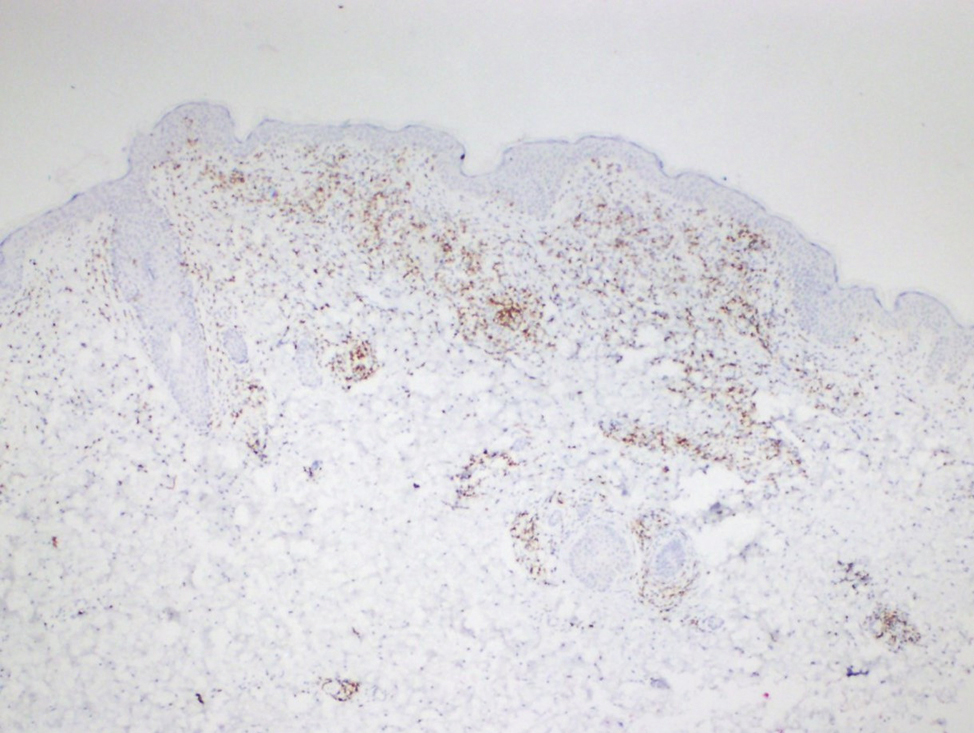
- Focal loss of CD7 expression (Diaminobenzidine, ×40)
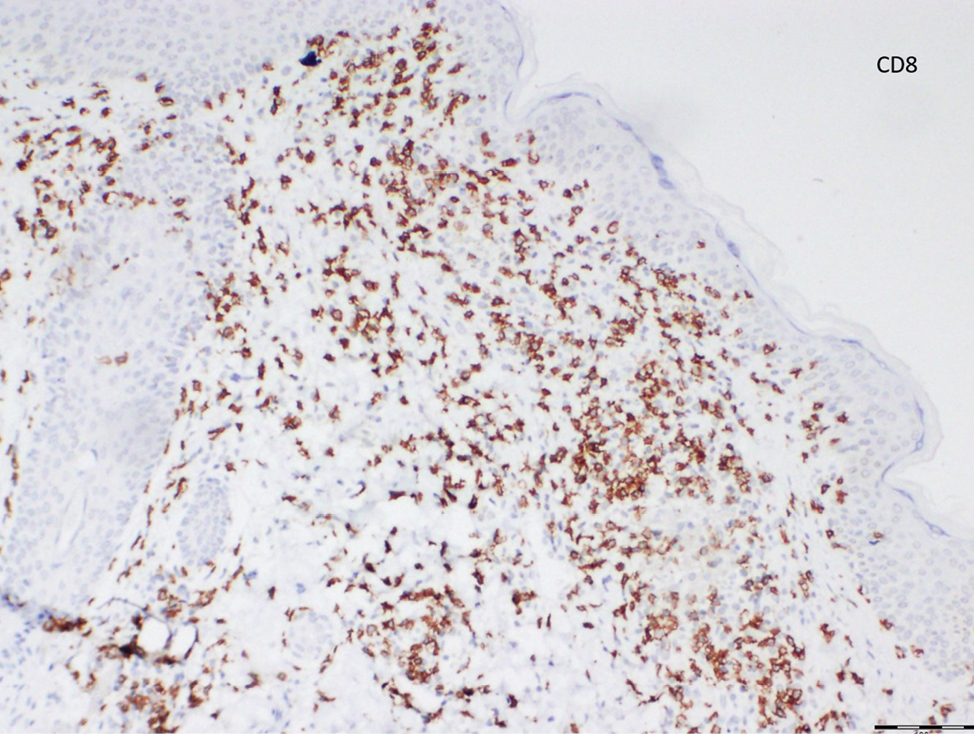
- The lymphocytes more profoundly express CD8 expression than CD4 expression (Diaminobenzidine, ×100)
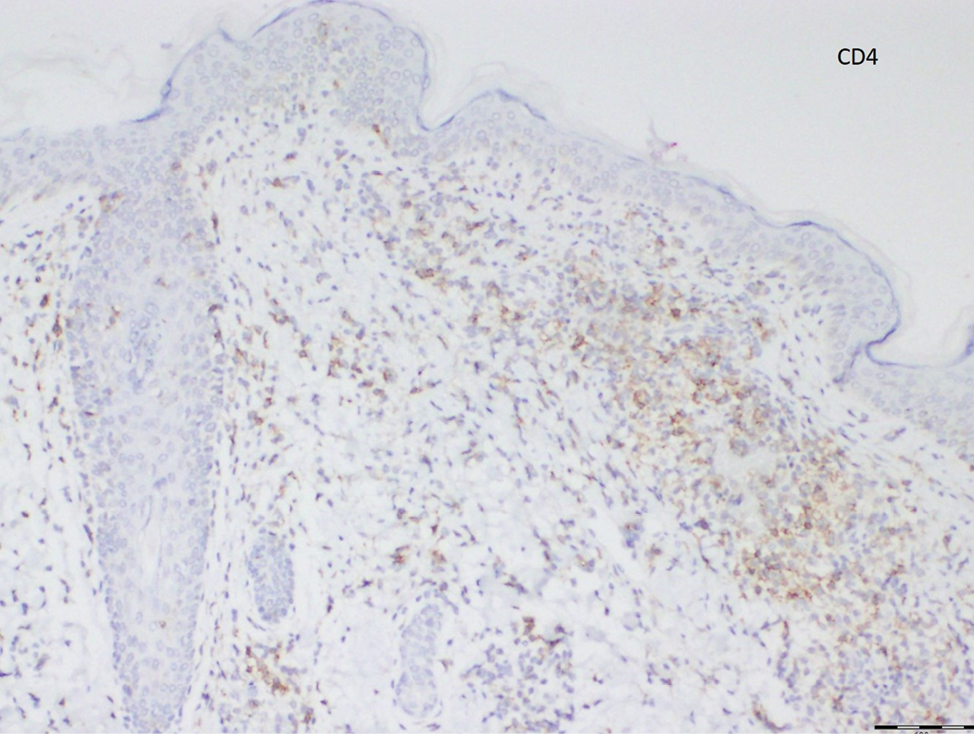
- The lymphocytes more profoundly express CD8 expression than CD4 expression (Diaminobenzidine, ×100)
Lichen aureus and unilesional mycosis fungoides were the main differential diagnoses in our patient. Because of considerable overlap, it is sometimes difficult to differentiate between pigmented purpuric dermatosis (PPD) and PPD-like mycosis fungoides clinically and histologically. It is well recognized that both disorders are much less common in children. Considering the progression of the lesion in a short time, a biopsy was performed on our patient. The presence of atypical lymphocytic infiltrates, microscopic evidence of diminished CD7 expression and clonal rearrangement in T-cell receptor gene arrangement study pointed to the probability of mycosis fungoides.
For many years the linkage between pigmented purpuric dermatosis and mycosis fungoides has been examined in three aspects.1 First, histologically pigmented purpuric dermatosis can mimic mycosis fungoides.2 Among its subtypes, lichen aureus is likely to be the strongest mimicker.3 Currently, pigmented purpuric dermatosis is phenotypically categorized into two types as polyclonal and monoclonal. Besides monoclonality, CD4+ mature T-cell dominance and loss of CD7 expression in pigmented purpuric dermatoses have been reported in the phenotypic and molecular studies.1,4,5 The presence of large cerebriform lymphocytes and Pautrier microabscesses, elevated CD4:CD8 (≥6) ratio and CD4 predominance is generally considered indicators of malignancy. Even so the absence of these findings does not exclude the diagnosis of mycosis fungoides. In our patient CD8 dominance was noted with CD7 loss. Fifteen per cent of mycosis fungoides show a reverse pattern characterized by CD8 dominance on microscopic examination.6 The lesion of our patient was very similar to the patient recently reported by Zubkov et al.7
Secondly, mycosis fungoides can present as PPD-like lesions.8 Autoimmune pathways are likely to be causative in mycosis fungoides related purpura.9 In a large series of patients in whom initial diagnosis was pigmented purpuric dermatosis, mycosis fungoides and suspected mycosis fungoides were detected in 8% (7/87) of patients.10 Notably, among six paediatric patients presenting with PPD-like lesions, one of them was diagnosed as mycosis fungoides. Magro et al. suggest performing T-cell rearrangement analyses in these patients with involvement of large surface areas who are more likely to exhibit monoclonality and loss of CD7 expression.4 Because of presence of CD7 loss, we performed T-cell receptor gene rearrangement study in our patient.
Lastly, mycosis fungoides may develop on preceding pigmented purpuric dermatosis.11,12 According to the literature lichen aureus should be regarded as ‘cutaneous T cell lymphoid dyscrasia’ with potential for progression to cutaneous T-cell lymphoma.4 In the study of Caytemel et al. 2 (1.5%) patients were evolved into mycosis fungoides.10 It is well documented that, only pigmented purpuric dermatosis with T-cell clonotype may progress to mycosis fungoides.4 On the contrary, some authors have proposed that classic lesions of lichen aureus do not progress to cutaneous T-cell lymphoma.13
In conclusion, our patient extraordinarily reflects the fact that the histopathological distinction of lichen aureus and mycosis fungoides can be blurred. Clinically solitary purpuric plaque with partial progression exhibiting exocytosis and nuclear atypia accompanied by CD7 loss, clonal proliferation and lack of characteristic microscopic findings of lichen aureus were strong arguments against the lichen aureus. However, as some authors, we believe that monoclonal lichen aureus belongs to the intermediate stage in the cutaneous T-cell dyscrasia spectrum. A long-term follow-up and serial biopsies are recommended. The question regarding categorizing these patients needs still to be addressed.
Declaration of patient consent
Patient’s consent not required as patient’s identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflict of interest
There are no conflicts of interest.
References
- Persistent pigmented purpuric dermatitis and mycosis fungoides: simulant, precursor, or both? A study by light microscopy and molecular methods. Am J Dermatopathol. 1997;19:108.:18.
- [CrossRef] [PubMed] [Google Scholar]
- Mimics of cutaneous lymphoma: report of the 2011 Society for Haematopathology/European Association for Haematopathology workshop. Am J Clin Pathol. 2013;139:536.:51.
- [CrossRef] [PubMed] [Google Scholar]
- Lichen aureus with pseudolymphomatous infiltrate. J Cutan Pathol. 2021;48:669.:73.
- [CrossRef] [PubMed] [Google Scholar]
- Pigmented purpuric dermatosis: classification by phenotypic and molecular profiles. Am J Clin Pathol. 2007;128:218.:29.
- [CrossRef] [PubMed] [Google Scholar]
- Pigmented purpuric dermatosis or mycosis fungoides: A diagnostic dilemma. Indian Dermatol Online J. 2016;7:183.:5.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Mycosis fungoides: A clinicopathological study of 60 cases from a Tertiary Care Center. Indian J Dermatol. 2020;65:123.:9.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Unilesional granulomatous pigmented purpuric dermatosis in a 7-year-old boy. Pediatr Dermatol. 2021;38:506.:7.
- [CrossRef] [PubMed] [Google Scholar]
- Mycosis fungoides presenting as pigmented purpuric dermatitis. Pediatr Dermatol. 2006;23:350.:4.
- [CrossRef] [PubMed] [Google Scholar]
- Pigmented purpura-like eruption as cutaneous sign of mycosis fungoides with autoimmune purpura. J Am Acad Dermatol. 1999;40:298.:9.
- [CrossRef] [PubMed] [Google Scholar]
- Pigmented purpuric dermatosis: Ten years of experience in a tertiary hospital and awareness of mycosis fungoides in differential diagnosis. J Cutan Pathol. 2021;48:611.:6.
- [CrossRef] [PubMed] [Google Scholar]
- Persistent pigmented purpuric eruption associated with mycosis fungoides: A case report and review of the literature. J Eur Acad Dermatol Venereol. 2001;15:62.:4.
- [CrossRef] [PubMed] [Google Scholar]
- Progression of pigmented purpura-like eruptions to mycosis fungoides: Report of three cases. J Am Acad Dermatol. 1988;19:25.:31.
- [CrossRef] [PubMed] [Google Scholar]
- Lichen aureus: clinicopathologic features, natural history, and relationship to mycosis fungoides. Arch Dermatol. 2008;144:1169.:73.
- [CrossRef] [PubMed] [Google Scholar]




