
Translate this page into:
Spontaneous splenic rupture: A rare complication in eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome)
*Corresponding author: Dr. Dharshini Sathishkumar, Department of Dermatology, Venereology and Leprology, Christian Medical College and Hospital, Vellore, Tamil Nadu, India. dharshsathish@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Shajil C, Sathishkumar D, George K, Thomas M. Spontaneous splenic rupture: A rare complication in eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome). Indian J Dermatol Venereol Leprol 2022;88:392-5.
Sir,
Eosinophilic granulomatosis with polyangiitis is a necrotising vasculitis affecting small- to medium-sized vessels of the skin, usually presenting with palpable purpura or subcutaneous nodules and rarely as urticaria, livedo reticularis or papulonecrotic lesions.1,2 The extracutaneous manifestations are commonly seen in the respiratory tract, peripheral nerves, gastrointestinal tract, kidneys and heart.2,3 Although splenic involvement is rare, we were unable to find any reports of spontaneous splenic rupture in eosinophilic granulomatosis with polyangiitis.3 Here, we report a case of eosinophilic granulomatosis with polyangiitis who had spontaneous splenic rupture 12 days after developing skin lesions.
A 33-year-old lady presented with painful blisters on the extremities for three days associated with fever and paraesthesia involving the feet. She had history of wheezing which improved with steroids and bronchodilators. There was no other history suggestive of connective tissue disease, loss of weight/appetite or drug intake. On examination, she had pedal oedema and multiple dull erythematous papules/plaques on the limbs with a dusky centre and a few also had central vesiculation [Figures 1a-d]. The differentials of cutaneous vasculitis, erythema multiforme and Sweet syndrome were considered and she was initiated on injection hydrocortisone and oral dicloxacillin. Histopathological examination of the skin lesion was consistent with small-vessel vasculitis with perivascular eosinophilic infiltrate [Figures 2a and b] and direct immunofluorescence showed immunoglobulin M deposition in a few superficial dermal blood vessels. The relevant laboratory investigations and imaging findings are shown in Table 1. A day after upper gastrointestinal endoscopy was done for the evaluation of trace blood in stools, she developed diffuse abdominal pain with swelling, tachycardia, cold peripheries, hypotension and persistent oozing from the site of intravenous cannulation.

- Multiple dull erythematous papules and plaques with dusky necrotic centre and a few with central vesicle/bulla on legs

- Multiple dull erythematous papules and plaques with dusky necrotic centre and a few with central vesicle/bulla on dorsum of hand

- Multiple dull erythematous papules and plaques with dusky necrotic centre and a few with central vesicle/bulla on dorsa of feet
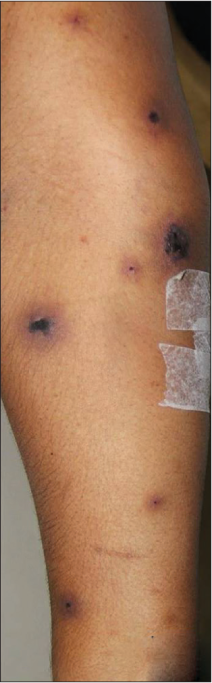
- Multiple dull erythematous papules and plaques with dusky necrotic centre and a few with central vesicle/bulla on right forearm

- Medium-vessel vasculitis with organising thrombi in deep dermis (haematoxylin and eosin, ×40)

- Perivascular scattered eosinophils (black arrow) (haematoxylin and eosin, ×200)

- Vasculitis with fibrinoid necrosis (black arrow) involving red pulp of spleen and perivascular eosinophilic infiltrate (blue arrow) (haematoxylin and eosin, ×100)
| Investigations | Results (normal range) |
|---|---|
| C-reactive protein | 109 mg/L (<6) |
| Total count and differential count 1 | 2,400 cells/mm3 (4000-12,000), neutrophils 59%, lymphocytes 16%, eosinophils 21% |
| Platelet count (at presentation) | 137,000 cells/mm3 (150,000–450,000) |
| Platelet count (day seven) | 44,000 cells/mm3 |
| Creatinine | 0.57 mg% (0.5–1.4) |
| Urine microscopy | Normal |
| Stool occult blood (two numbers) | Trace |
| Immunoglobulin E | 12,673.3 U/ml (5–100) |
| ASO† | <50 IU/ml (<300) |
| Cold agglutination | Negative |
| Blood culture | No growth |
| ANA‡ | Negative |
| C3/C4 | 118/38.3 mg/dl (90–180/10–40) |
| Anti-PR3§ (c-antineutrophil cytoplasmic antibody)/anti-myeloperoxidase (p-antineutrophil cytoplasmic antibody) | <2 U/ml (<20)/ 4.2 U/ml (<20) |
| Rheumatoid arthritis factor | <8.8 IU/ml (<20) |
| Cryoglobulins | Not detected |
| Anti-prothrombin phosphatidylserine complex antibody | 4 U/ml (<16) |
| Anticardiolipin antibody | 2 GPL (<20) |
| Lupus anticoagulant | Negative |
| Antibodies to β2 glycoprotein complex immunoglobulin M/immunoglobulin G | 6/2 Ru/ml (<20/<20) |
| Anti-SSA antibodies | 1 Ru/ml (<20) |
| Homocysteine estimation | 6.1 umol/L (3.7–13.9) |
| Chest X-ray and computed tomography thorax | Well-defined 4×3 mm calcified soft-tissue nodule in the left lung (sequela of past infection/inflammation) |
| Nerve conduction study | Sensorimotor axonal neuropathy involving the lower limbs |
Laboratory investigations showed a drop in haemoglobin (from 11 g/dl to 4.2 g/dl) and prolonged coagulation parameters, suggesting disseminated intravascular coagulation. The patient was resuscitated and rushed to the operation theatre considering the possibility of an intraperitoneal bleed. Intraoperative upper gastrointestinal endoscopy ruled out gastrointestinal perforation and emergency laparotomy revealed massive haemoperitoneum with mild splenomegaly and stellate rupture of the spleen. Splenectomy was done and histopathological examination of the splenic tissue showed vasculitis with eosinophilic infiltrate [Figure 2c]. After surgery, the patient gradually recovered with antibiotics (meropenem, metronidazole) and other supportive measures. The diagnosis of eosinophilic granulomatosis with polyangiitis was made according to the American College of Rheumatology criteria.1 Her skin lesions showed remarkable improvement [Figure 3] on treatment with prednisolone (0.6 mg/kg) and mycophenolate mofetil. The disease has been in remission for the past 2.5 years and she is currently on low-dose prednisolone (5 mg) and mycophenolate mofetil.

- Lesions have completely healed with post-inflammatory hyperpigmentation (2.5 years follow-up)
Eosinophilic granulomatosis with polyangiitis is an antineutrophil cytoplasmic antibody-associated vasculitis and its natural course is divided into three phases. The first phase is characterised by allergic rhinosinusitis and adult-onset asthma followed by peripheral blood eosinophilia with end-organ eosinophilic infiltrate in phase two. Systemic vasculitis with granulomatous inflammation in phase three can lead to complications such as myocardial infarction, heart failure, gastrointestinal perforation and venous thromboembolism.1,2
The American College of Rheumatology criteria proposed in 1990 include asthma, eosinophilia, neuropathy, sinusitis, non-fixed pulmonary infiltrate and eosinophilic infiltrate in the biopsy. The presence of four out of six criteria diagnoses the condition with 85% sensitivity and 99.7% specificity.1,2 Our patient had cutaneous small-vessel vasculitis with eosinophilic infiltrate, asthma, peripheral eosinophilia and peripheral neuropathy, thus fulfilling the criteria. Her raised immunoglobulin E level also favoured the diagnosis. Although extravascular red granulomas are described in eosinophilic granulomatosis with polyangiitis, it is neither a pathognomonic feature nor mandatory for diagnosis.2,3 Antineutrophil cytoplasmic antibody positivity, absent in this case, is not a prerequisite for diagnosis, as it is seen in only 40–60% eosinophilic granulomatosis with polyangiitis cases.1,2 The prognosis depends on the severity of the disease, with studies demonstrating five-year survival rate of 88.9% and ten-year relapse-free survival rate of 54.4%.1,2
The occurrence of spontaneous splenic rupture makes this case unique. Spontaneous splenic rupture is a rare morbid condition, causes of which fall into six major groups: neoplastic, infectious, inflammatory, structural, iatrogenic and idiopathic.4,5 It is reported in various rheumatologic conditions such as rheumatoid arthritis, systemic lupus erythematosus, polyarteritis nodosa with greater frequency and in granulomatosis with polyangiitis infrequently.5 It presents with left upper abdominal pain, abdominal distension, tenderness and guarding. Ultrasound helps in rapid diagnosis with exploratory laparotomy being both therapeutic and diagnostic. Although the predisposing factors for spontaneous splenic rupture have not been clearly defined, McCain et al. have proposed splenic vasculitis, vessel destruction and necrosis as the possible triggers.4,5 In our case, spontaneous splenic rupture was probably secondary to splenic vasculitis as evident on histopathological examination. She was vaccinated to prevent overwhelming post-splenectomy infections, a fatal long-term complication following splenectomy.
Even though spontaneous splenic rupture is a rare complication of vasculitis, it should be considered in a patient with vasculitis presenting with features of with acute abdomen, as timely diagnosis and intervention improve the overall outcome as evident in our case.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Eosinophilic granulomatosis with polyangiitis: An overview. Front Immunol. 2014;5:549.
- [CrossRef] [PubMed] [Google Scholar]
- Eosinophilic granuloma and necrotizing vasculitis (Churg-Strauss syndrome) Involving a parotid gland, lymph nodes, liver and spleen. Scand J Rheumatol. 1989;18:171-5.
- [CrossRef] [PubMed] [Google Scholar]
- Life-threatening spontaneous splenic rupture with systemic lupus erythematosus: Case report and literature review. Clin Rheumatol. 2012;31:1019-25.
- [CrossRef] [PubMed] [Google Scholar]
- Splenic rupture as the presenting manifestation of vasculitis. Semin Arthritis Rheum. 2002;31:311-6.
- [CrossRef] [PubMed] [Google Scholar]




