Translate this page into:
Subcutaneous panniculitis-like T-cell cutaneous lymphoma
2 Department of Dermatology, Safdarjung Hospital, New Delhi, India
Correspondence Address:
Avninder Singh
Institute of Pathology-ICMR, Safdarjung Hospital Campus, New Delhi - 110 029
India
| How to cite this article: Singh A, Kumar J, Kapur S, Ramesh V. Subcutaneous panniculitis-like T-cell cutaneous lymphoma. Indian J Dermatol Venereol Leprol 2008;74:151-153 |
Abstract
Subcutaneous panniculitis-like T cell lymphoma (SPTCL) is a rare cytotoxic T-cell lymphoma classified in the World Health Organization-European Organization for Research and Treatment of Cancer (WHO-EORTC) classification as a unique extranodal lymphoma with characteristic by T cell receptor (TCR) gene rearrangement. We report here a case of SPTCL in a 22 year-old woman who had presented with variably sized multiple nodules on both her legs. Initial differential diagnoses considered were panniculitis and lupus panniculitis. The histopathology showed a predominantly subcutaneous lobular infiltrate with atypical lymphocytes, karyorrhexis and rimming of adipocytes by lymphoid cells. Immunohistochemistry showed CD4−, CD8+, CD56− T-cell phenotype. Although TCR rearrangement studies were not done, the above T-cell phenotype and sparing of epidermis and dermis suggested the possibility of an SPTCL α/β type. The patient received five cycles of a cyclophosphamide, doxorubicin, vincristine and prednisone (CHOP) regimen which resulted in the regression in her skin lesions and constitutional symptoms. |
| Figure 3: Photomicrograph showing: (A) diffuse lymphoid infiltrate in subcutaneous fat (H and E, �40), (B) karyorrhexis and atypical lymphocytes rimming the fat cells (H and E, �400) |
|
| Figure 3: Photomicrograph showing: (A) diffuse lymphoid infiltrate in subcutaneous fat (H and E, �40), (B) karyorrhexis and atypical lymphocytes rimming the fat cells (H and E, �400) |
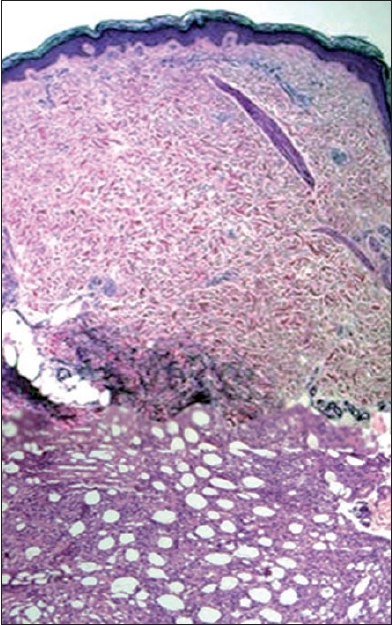 |
| Figure 2: Photomicrograph showing predominant subcutaneous infiltrate with sparing of epidermis and dermis (H and E, �40) |
|
| Figure 2: Photomicrograph showing predominant subcutaneous infiltrate with sparing of epidermis and dermis (H and E, �40) |
 |
| Figure 1: Annular plaque on the thigh showing papulonodules, central depression, peripheral erythema and induration |
|
| Figure 1: Annular plaque on the thigh showing papulonodules, central depression, peripheral erythema and induration |
Introduction
Subcutaneous panniculitis-like T-cell lymphoma (SPTCL) is an uncommon extranodal lymphoma involving the skin that can mimic other panniculitic conditions. Its prognosis is bad so it is important to exclude other benign panniculitic conditions especially lupus panniculitis and erythema nodosum. Only two cases of SPTCL have been reported from India, the first describing its cyto-histological findings and response to chemotherapy while the second was rapidly fatal. [1],[2] SPTCL is rare but it is important for dermatologists and pathologists to be aware of this entity due to its clinical and histological resemblance to lupus panniculitis.
Case Report
A 22 year-old woman presented with variably sized multiple nodules seen on both her legs for the last ten months along with malaise and loss of appetite for the last two months. These nodules first appeared on the thigh, were about 0.5 cm in size and slowly grew larger by peripheral extension to form annular plaques. On physical examination, the initial lesion revealed an annular plaque measuring 7 x 6 cm with a central depression, peripheral erythema and induration and was studded with multiple papulo-nodules that were firm and nontender [Figure - 1]. Other lesions were smaller, ranging from 1 x 1 to 3 x 3 cm. There was no palpable lymphadenopathy, peripheral nerve thickening or sensory involvement and systemic examination was unremarkable. Laboratory investigations showed a normal hemogram and normal liver and renal function tests. Chest X-ray was also normal. As the patient hailed from a leprosy-endemic region, leprosy in reaction, panniculitis and lupus panniculitis were considered as differential diagnoses. Antinuclear antibody titers were normal and antibodies to double-stranded DNA were absent.
Histopathological examination of the skin biopsy taken from the periphery of the initial lesion showed a lymphomatous infiltrate involving predominantly the subcutaneous lobules in a lobular panniculitis-like pattern. The epidermis and dermis were spared by this infiltrate [Figure - 2]. On higher magnification, the lymphoid infiltrate was seen to be diffuse [Figure - 3]A and had enlarged and hyperchromatic nuclei, occasional mitoses, karyorrhexis and rimming of the adipocytes by these atypical neoplastic lymphoid cells [Figure - 3]B. Phagocytosis of this karyorrhectic nuclear debris by histiocytes was also seen. Immunohistochemical evaluation showed CD3, CD45 RO and CD8 positivity and CD20, CD4, CD30 and CD56 negativity. Neither a T cell receptor (TCR) gene rearrangement study nor a bone marrow examination was done in this case. Although definite proof is lacking, our case most probably represents an SPTCL α/β T-cell phenotype given the strictly subcutaneous localization and the CD4− , CD8 + and CD56− immunophenotyping. The patient was referred to the medical oncology department with a diagnosis of SPTCL. She underwent five cycles of a CHOP chemotherapy regimen (cyclophosphamide 900 mg, doxorubicin 70 mg, vincristine 2 mg on day one along with oral prednisolone). Therapy in each cycle lasted for five days with a gap of 16 days. The patient was hospitalized after the first cycle for fever and vomiting. She was treated with subcutaneous granulocyte-colony stimulating factor (G-CSF) and supportive measures. Six months after the initial diagnosis, the lesions were found to be softened and the induration had regressed. The patient is under regular follow-up and her appetite has marginally improved.
Discussion
SPTCL is a rare cytotoxic T-cell lymphoma of the skin and accounts for less than 1% of all non-Hodgkin′s lymphoma cases. [3] It was originally described by Gonzalez et al. [3] in 1991 as an uncommon variant of lymphoma that is localized to the subcutis and mimics lupus panniculitis. It was proposed as a provisional clinicopathologic entity by the EORTC cutaneous lymphoma classification [4] and later incorporated into the WHO classification of hematopoietic and lymphoid tumors. [5] Clinically, they present as multiple plaques or nodules on the extremities and the trunk with almost equal gender distribution. The mean age at diagnosis is 39 years; constitutional symptoms are seen in 40% of the cases and an association with hemophagocytic syndrome (HPS) is seen in 45% of the cases. [6] Histopathological findings reveal lobular panniculitis-like infiltration by atypical lymphoid cells, which are seen rimming the adipocytes in a lace-like pattern. Nuclear pleomorphism, mitoses, karyorrhexis and hemophagocytosis by histiocytes with ′bean-bag′ appearance are usually seen. Immunohistochemical studies have demonstrated that these atypical neoplastic lymphoid cells have a cytotoxic T-cell phenotype. [7] Most of the atypical lymphoid cells express CD3, CD8, T-cell intracellular antigen (TIA-1) and perforin and lack CD4, CD30 and CD56 expression. TCR gene analysis by polymerase chain reaction-single strand conformational polymorphism divides SPTCL into two subtypes: (1) those derived from T-cells expressing TCR-α/β and normally CD8+ and (2) those derived from γ/δ T-cells and commonly expressing the NK cell marker, CD56. [8]
SPTCL must be differentiated from other benign causes of lobular panniculitis, like lupus erythematosus profundus and Weber-Christian disease that may be clinically and histologically confused with SPTCL. The absence of basal layer liquefaction and the presence of atypical lymphoid cells, mitosis, tissue necrosis will help morphological differentiation. Similarly, other differential diagnoses may include cutaneous T-cell lymphomas like NK/T cell lymphomas or anaplastic large cell lymphomas. The former shows the extension of a neoplastic infiltrate into the dermis and epidermis along with the presence of EB virus sequences, angioinvasion and CD56 positivity, while the latter shows epidermal ulceration and CD30 positivity.
The treatment of SPTCL is not standardized and may include systemic steroids, multidrug chemotherapy or cyclosporine. The response is variable but generally the prognosis is poor in the presence of constitutional symptoms, cytopenia, involvement of multiple sites and associated hemophagocytic syndrome (HPS). The increased proportion of γ/δ subsets and expression of CD56 are more prevalent among patients who develop HPS and have progressive disease with a mortality rate of over 50%. [6] Recent studies have shown that clinical, histological and immunophenotypical differences between cases of SPTCLs with α/β T-cell phenotype and those with γ/δ T-cell phenotype, suggesting that they may be different entities. [9] In a more recent review of the WHO-EORTC classification of cutaneous lymphomas, Willemze et al. [10] have suggested that the term ′SPTCL′ should be restricted to cases with α/β+ T-cell phenotype and that cases with γ/δ+ T-cell phenotype be called cutaneous γ/δ+ T-cell lymphomas.
In conclusion, SPTCL is a distinct variant of a lymphoma derived from either α/β or γ/δ T cells. The former tends to follow a more indolent course while the latter is more aggressive with a poor clinical outcome. The prudent utilization of molecular techniques like TC rearrangement helps to predict which SPTCL requires more aggressive treatment from the outset.
| 1. |
Goel K, Kini H, Rau AR, Nadar S, Pai MR, Rao HT. Cytomorphology of subcutaneous panniculitic T-cell lymphoma (SPTCL): A case report. Indian J Pathol Microbiol 2006;49:246-8.
[Google Scholar]
|
| 2. |
Bandyopadhyay SK, Dutta SK, Dutta A. Panniculitis-like T-cell lymphoma with fatal termination. J Indian Med Assoc 2005;103:551-2.
[Google Scholar]
|
| 3. |
Gonzalez CL, Medeiros LJ, Braziel RM, Jaffe ES. T-cell lymphoma involving subcutaneous tissue: A clinicopathologic entity commonly associated with hemophagocytic syndrome. Am J Surg Pathol 1991;15:17-27.
[Google Scholar]
|
| 4. |
Willenze R, Kerl H, Sterry W, Berti E, Ceronni L, Chimenti S, et al. EORTC classification for primary cutaneous lymphomas: A proposal from the Cutaneous Lymphoma Study Group of the European Organization for Research and Treatment of Cancer. Blood 1997:90:354-71.
[Google Scholar]
|
| 5. |
Jaffe ES, Harris NL, Stein H, Vardiman JW. WHO classification of tumors. In : Pathology and Genetics of Tumors of Hemopoeitic and Lymphoid tissues . Lyon: IARC Press; 2001.
[Google Scholar]
|
| 6. |
Weenig RH, Ng CS, Perniciaro P. Subcutaneous panniculitis-like T-cell lymphoma: An elusive case presenting as lipomembranous panniculitis and a review of 72 cases in the literature. Am J Dermatopathol 2001;23:206-15.
[Google Scholar]
|
| 7. |
Kumar S, Krenacs L, Medeiros J, Elinitoba-Johnson KS, Greiner TC, Sorbara L, et al. Subcutaneous panniculitic T-cell lymphoma is a tumor of cytotoxic T lymphocytes. Hum Pathol 1998;29:397-403.
[Google Scholar]
|
| 8. |
Salhany KE, Macon WR, Choi JK, Elinitas L, Lessin SR, Felgar RE, et al. Subcutaneous panniculitic-like T-cell lymphoma: Clinicopathologic, immunophenotypic and genotypic analysis of alpha/beta and gamma/delta subtypes. Am J Surg Pathol 1998;22:881-3.
[Google Scholar]
|
| 9. |
Santucci M, Pimpinelli N, Massi D, Kadin ME, Meijer CJ, Muller-Hermelink HK, et al. Cytotoxic/NK cell lymphomas: Report of EORTC cutaneous lymphoma task force workshop. Cancer 2003;97:610-27.
[Google Scholar]
|
| 10. |
Willemze R, Jaffe ES, Burg G, Ceronni L, Berti E, Swerdlow SH, et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005;105:3768-85.
[Google Scholar]
|
Fulltext Views
4,245
PDF downloads
1,045





![[Figure - 1]](#fig_ijdvl_2008_74_2_151_39703_1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2008_74_2_151_39703_2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2008_74_2_151_39703_3.jpg){kind=link}