
Translate this page into:
Systemic sarcoidosis during pazopanib treatment for clear cell renal cell carcinoma
Corresponding author: Dr. Ilenia Marafioti, Via C. Valeria, Gazzi. 98125 Messina, Italy. ileniamarafioti@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Vaccaro M, Borgia F, Lentini M, Gaeta M, Marafioti I. Systemic sarcoidosis during pazopanib treatment for clear cell renal cell carcinoma. Indian J Dermatol Venereol Leprol 2023;89:444-6.
Dear Editor,
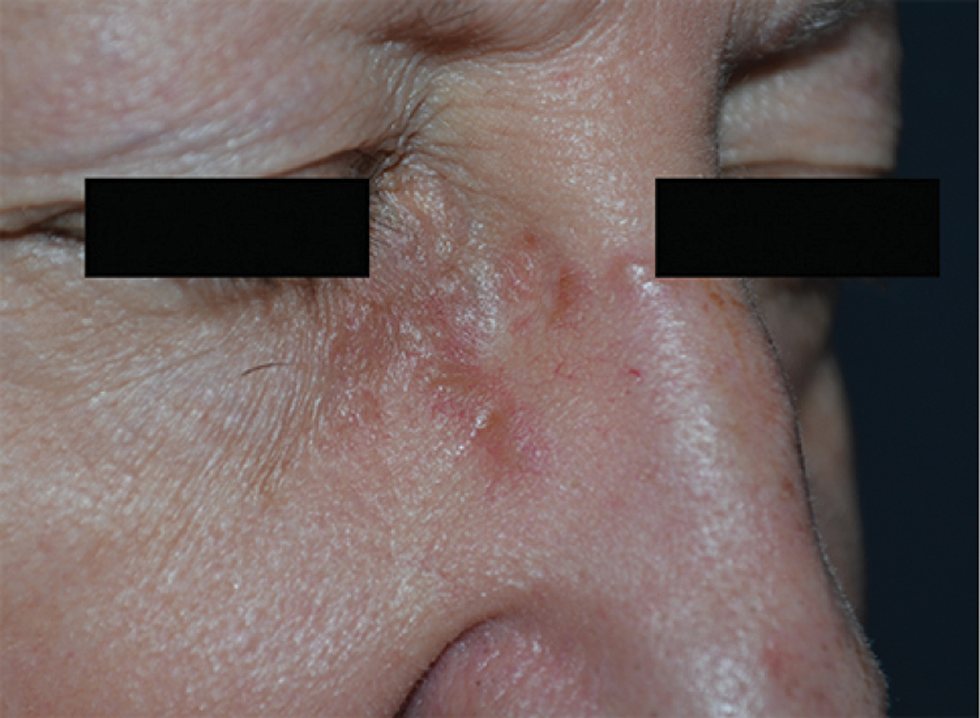
Sarcoidosis is a multi-organ disease of unknown aetiology, that preferentially affects middle-aged females. It can manifest in almost any organ, such as the lungs, heart, skin and eyes. Cutaneous lesions occur in about 30% of sarcoid patients, presenting as papules, nodules, plaques, ulcers and alopecia.1 The association with malignancies has already been reported. The diagnosis of sarcoidosis may precede or occur after malignancy. It can have a paraneoplastic origin or it can be triggered by antineoplastic treatments.2 Here we present a case of a 64-year-old woman who was referred to our department for the appearance of skin lesions. Approximately 12 years ago, she was diagnosed with clear cell renal cell carcinoma, Fuhrman grade II. Eight years later, vertebral metastases were found. Given the poor response to radiotherapy, tyrosine kinase inhibitor, pazopanib was started. After two years of treatment, a computer tomography scan revealed two millimetric nodules in the inferior lobe of the right lung, without lymphadenopathy. These lesions showed a slight increase in size at one-year follow-up, together with the appearance of similar new lesions in the middle and upper right lobes and multiple mediastinal lymphadenomegaly. “Tree in bud” parenchymal thickening was also detected [Figure 1]. Over a period of six months, imaging techniques highlighted a progressive involvement of the lungs and the onset of micronodular splenomegaly. Positron emission tomography showed hypermetabolic activity in the lungs, spleen and several lymph nodes (including supraclavicular, mediastinal and inguinal ones). Mediastinal lesions were partially responsive to systemic steroids; hence a supraclavicular lymph node biopsy was performed. Histology showed epithelioid granulomatous lymphadenopathy, without necrosis. After three months, several asymptomatic coalescing papules appeared on the forehead, on the root of the nose and on the posterior neck region. They were yellowish in colour with irregular surfaces, ill-defined borders and an overall diameter between 15 and 25 mm. Some thin vessels were visible to the naked eye [Figures 2a and b]. Dermoscopy revealed structureless yellow-orange areas, well-focused linear irregular vessels and dilated follicles [Figure 2c]. Histology confirmed the diagnostic suspicion of sarcoidosis [Figures 3a and b]. Because of the rapid worsening of the general condition, the patient was lost at follow-up. We eventually found out about the patient’s demise.
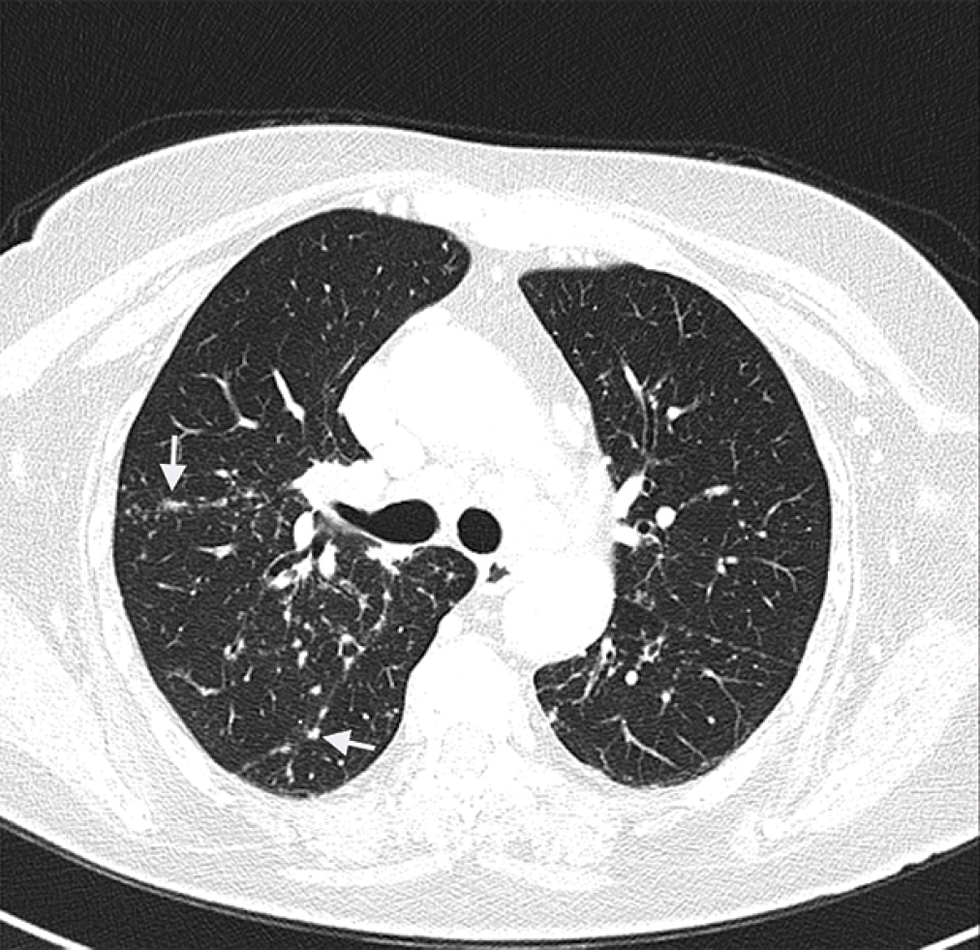
- Multiple small sub-pleural, peri-bronchovascular and centrilobular nodules (arrows) in the lungs with multiple mediastinal lymphadenopathies (CT scan)
- Structureless orange areas with well-focused linear irregular vessels and dilated follicles on dermoscopy (×20)
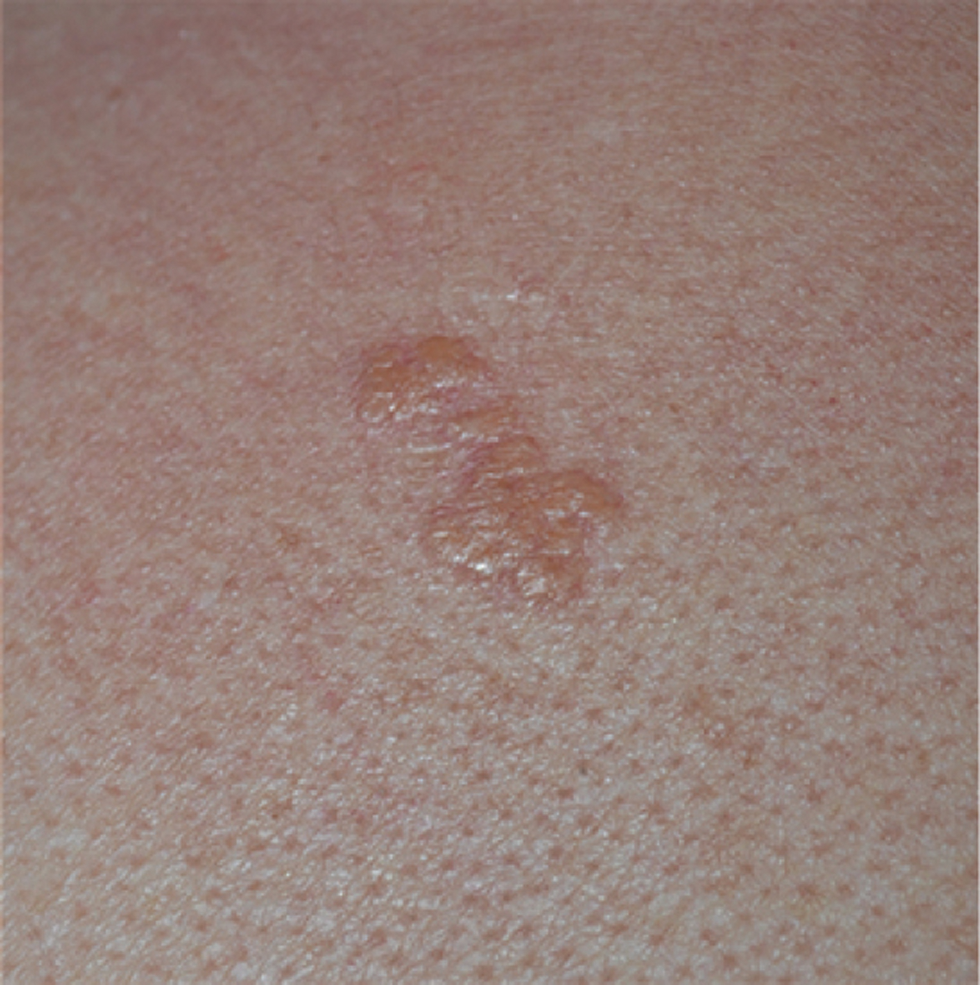
- Sarcoidosis skin lesions on the back of the neck
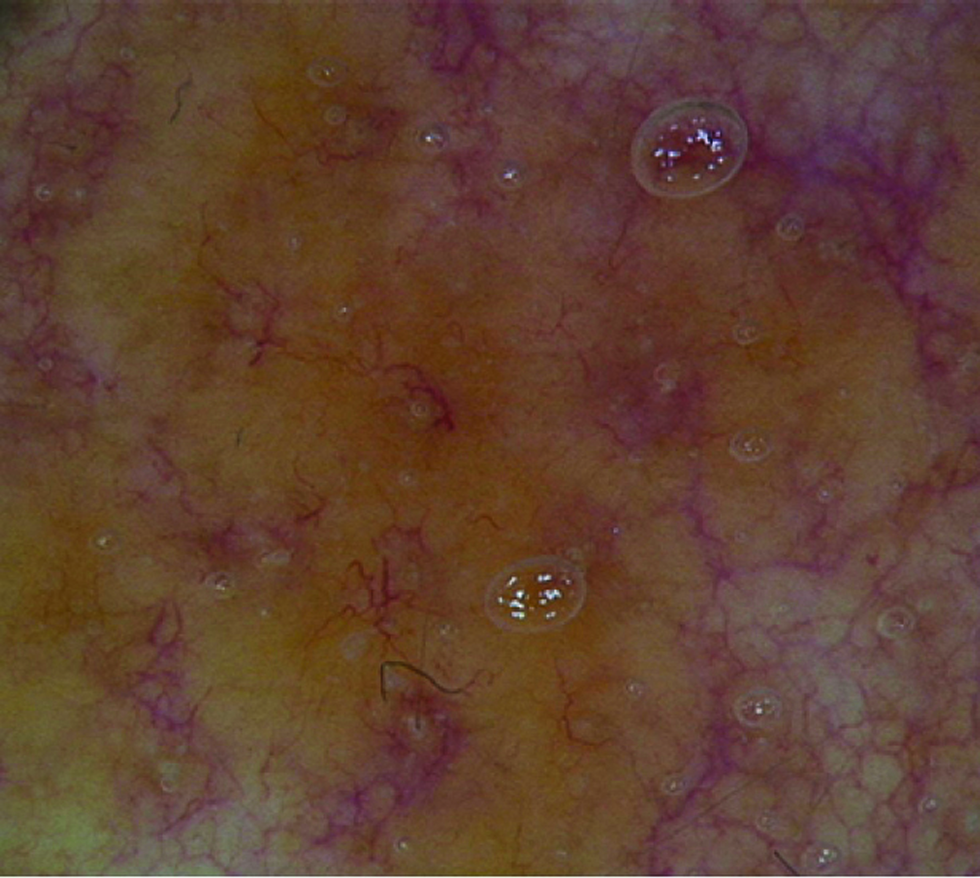
- Dermoscopy showed structureless yellowish-orange areas and well-focused linear irregular vessels (×20)
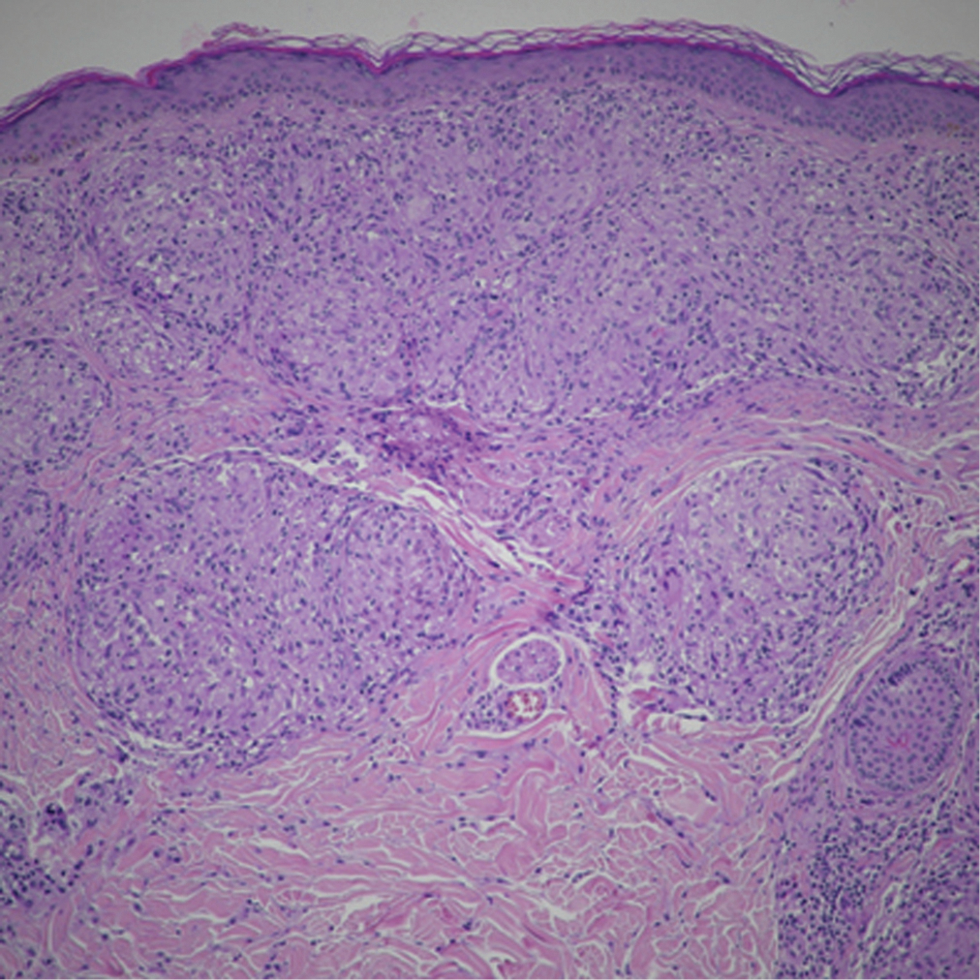
- Sharply defined “naked” sarcoidal granulomas with sparse few lymphocytes and no necrosis (H&E, ×200)
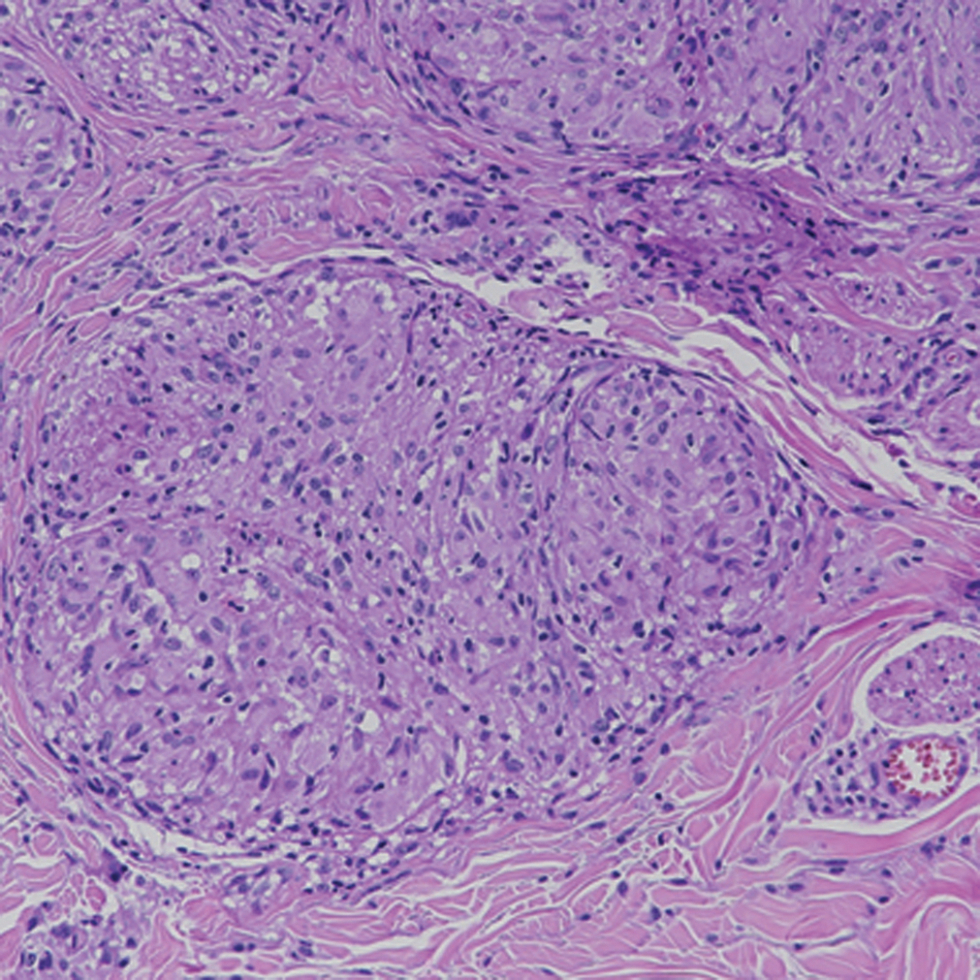
- “Naked” granuloma is sharply defined with sparse lymphocytes and no necrosis, consistent with sarcoidal granuloma (H&E, ×400)
The association between cancer and sarcoidosis is well known. Linkage analysis revealed that sarcoidosis and malignancies may be aetiologically related in at least 25% cases.3 Sarcoidosis-related tumours include haematologic malignancies and solid cancers (e.g., liver, lung, skin, testicle, cervix and uterus). Reports about sarcoidosis and renal cell carcinoma are very limited. Granulomatous reactions associated with clear cell renal cell carcinoma in literature are mainly represented by sarcoid-like granulomas, non-necrotizing collections of epithelioid cells found in oncologic patients not fulfilling diagnostic criteria for systemic sarcoidosis.4 They occur in 4.4% of carcinomas, 13.8% of Hodgkin and 7.3% of non-Hodgkin lymphomas.5 Sarcoid-like granulomata (SLGs) can arise in regional draining lymph nodes, spleen, bone marrow and skin but also in both primary or metastatic neoplastic tissue. They have been interpreted as a T-cell response to antigenic factors released by tumour cells during necrosis.6 The distinction between sarcoidosis and sarcoid-like granuloma is important in establishing the risk of systemic disease. They both show fluorodeoxyglucose avidity mimicking malignancy dissemination, so histologic confirmation is always needed. Sarcoid-like granulomas differ from sarcoidosis granulomas by the presence of B-cells, prominent sinus histiocytosis and poor fibrosis. In our patient, the onset of lung disease appeared after two years’ of treatment with pazopanib. It is known that some antineoplastic drugs, including pembrolizumab, ipilimumab, nivolumab, interferon and interleukin-2, can induce or exacerbate sarcoidosis, especially in patients with haematologic malignancies. But until now, pazopanib has not been reported as a causative drug.2 Moreover, its mechanism of action does not explain the onset of the disease. Discontinuation of chemotherapy should be useful to distinguish a drug-related reaction from a disease-related one, avoiding biopsy. Unfortunately, this is often not possible since life-saving drugs are involved. Our case showed a very rare association between clear cell renal cell carcinoma and systemic sarcoidosis in the course of treatment with pazopanib. Although it is not possible to establish with certainty whether sarcoidosis was primary or secondary to the tumour or its treatment, our experience seemed worthy of note. Supported by the literature data in oncologic patients, an aetiological correlation between sarcoidosis and the underlying renal carcinoma seems the most likely hypothesis, while treatment with pazopanib is probably temporal rather than causally related to the systemic disease. However, the rapid progression of granulomatous manifestations after pazopanib therapy, does not allow to exclude its negative influence on the course of sarcoidosis.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflict of interest
There are no conflicts of interest.
References
- Linkage analysis of malignancy-associated sarcoidosis. Chest. 1995;107:605-13.
- [CrossRef] [PubMed] [Google Scholar]
- The Japanese Society of Sarcoidosis and Other Granulomatous Disorders (JSSOG) Diagnostic Standard and Guideline for Sarcoidosis-2015 <http://jssog.com/www/top/shindan/shindan2-1new.html>
- Sarcoid reactions in malignant tumours. Cancer Treat Rev. 1986;13:147-56.
- [CrossRef] [PubMed] [Google Scholar]
- Sarcoid reactions in primary pulmonary carcinoma: Report of seven cases. Oncol Rep. 1998;5:177-80.
- [PubMed] [Google Scholar]




