Translate this page into:
Two novel mutations of the ADAR1 gene in Chinese patients with dyschromatosis symmetrica hereditaria
2 Shandong Provincial Hospital for Skin Diseases, Shandong University; Shandong Provincial Medical Center for Dermatovenereology, Jinan, Shandong, China
3 Shandong Provincial Institute of Dermatology and Venereology, Provincial Academy of Medical Science; Shandong Provincial Hospital for Skin Diseases, Shandong University; Shandong Provincial Key Lab for Dermatovenereology; Shandong Provincial Medical Center for Dermatovenereology, Jinan, Shandong, China
Correspondence Address:
Furen Zhang
Shandong Provincial Institute of Dermatology and Venereology, 27397. Jingshi Road, Jinan, Shandong Province, 250 022
China
| How to cite this article: Yuan C, Liu H, Fu X, Yu Y, Yu G, Bao F, Lu N, Li J, Liu J, Tian H, Zhang F. Two novel mutations of the ADAR1 gene in Chinese patients with dyschromatosis symmetrica hereditaria. Indian J Dermatol Venereol Leprol 2012;78:746-748 |
Sir,
Dyschromatosis symmetrica hereditaria (DSH [MIM127400]) is an autosomal dominantly inherited disease, characterized by a mixture of hyperpigmented and hypopigmented macules on the face and the back of the extremities; however, sporadic cases have also been reported. Heterozygous mutations acting on double-stranded RNA-specific adenosine deaminase (ADAR1 or DSRAD) gene were identified as the molecular basis of DSH. [1] So far, about 121 different mutations of this gene have been reported. Here, we report 2 novel and 1 recurrent mutation of the ADAR1 gene in sporadic Chinese patients with DSH.
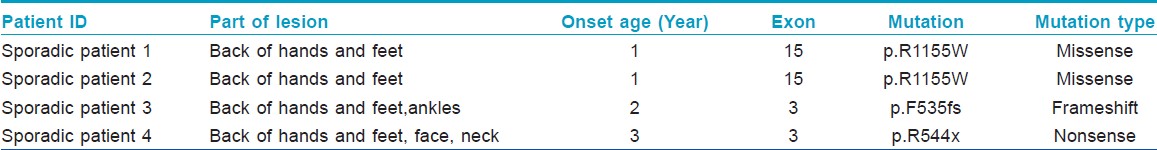
In this study, we investigated 4 sporadic cases that had no positive family histories from the Shandong Province of China. All the cases have a mixture of hyperpigmented and hypopigmented macules on the back of hands and feet [Figure - 1]a-d. All the clinical and molecular findings are summarized in [Table - 1].
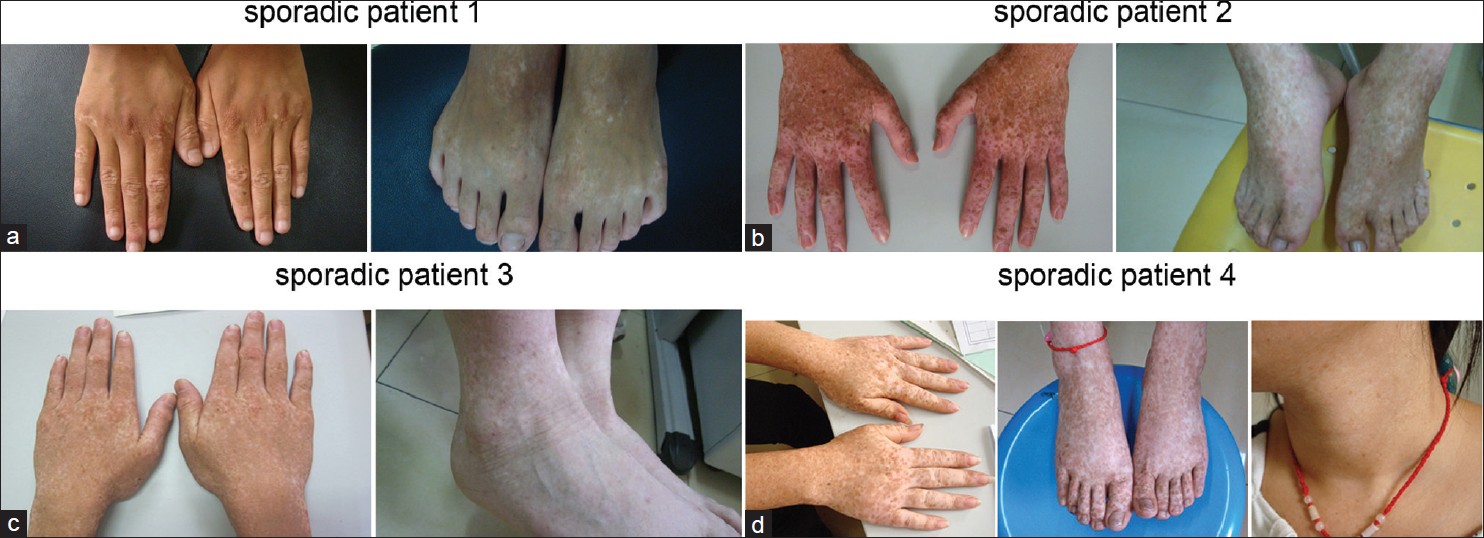 |
| Figure 1: (a-d) Clinical appearance of the 4 sporadic cases |
After informed consent and approval of the ethics committee of the institute, genomic DNA was extracted from the peripheral blood of the 4 cases and 100 normal healthy Chinese people. All the 15 exons of ADAR1 genes and their flanking intronic sequences of 200 bps were amplified by polymerase chain reaction. Products were purified and directly sequenced on ABI 3130 x l Genetic Analyzer.
We identified 2 novel mutations (p.F535fs-563x, p.R544X) and 1 recurrent missense mutation (p.R1155W) [Figure - 2]. None of these mutations was found in 100 controls. Mutations were identified by comparing with the reported cDNA reference sequence (GenBank accession number: NM_001111).
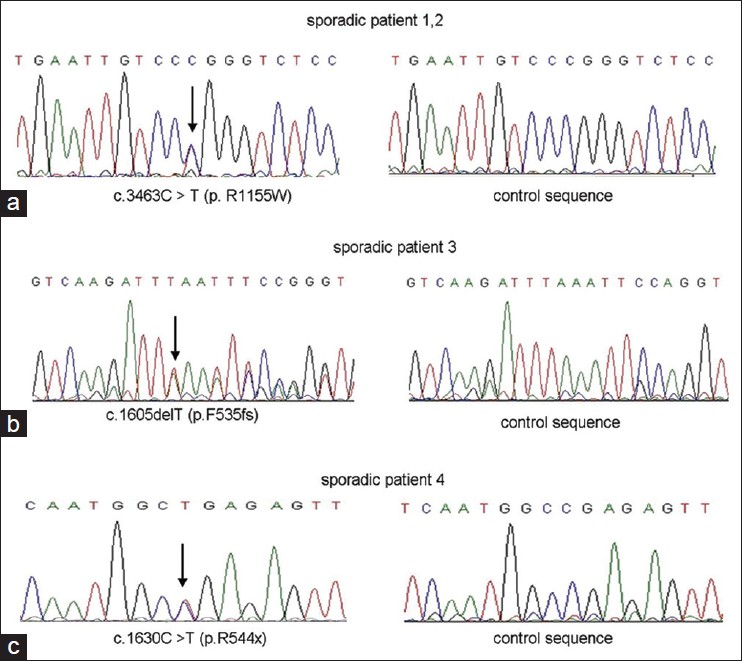 |
| Figure 2: (a) 3463C > T (p.R1155W) missense mutation. (b) 1605delT (p.F535fs) framshift mutation. (c) Nonsense mutation c.1630C > T (p.R544x) |
The human ADAR1 gene encodes RNA-specific adenosine deaminase and contains 15 exons. The enzyme has 2 Z-alpha domains (Z-alpha), 3 dsRNA binding domains (DSRM) and the putative deaminase domain (ADEAMc), corresponding to exon 2, exons 2-7, and exons 9-15 of ADAR1, respectively. So far, a total of 121 different mutations of the ADAR1 gene have been reported with DSH. Sixty five of the 121 mutations were located within ADEAMc domain. This domain is critical for enzyme function and is thought to play an important role in the pathogenesis of DSH. The missense mutation 3463C > T (p.R1155W) which was identified in the sporadic case 1, 2 is located in the deaminase domain of the ADAR1 protein in exon 15. It has also been reported in another 3 unrelated families, [2],[3],[4] and this result further supported the speculation that the deaminase domain might be a hot spot for mutations.
The c.1605 del T (p.F535fs) mutation was found in the sporadic patient 3. The base "T" deletion caused a frameshift mutation with the change of amino acids from 535 to 562 and introduced a new terminating TGA codon at 28 codons downstream of deletion site at position 563. The nonsense mutation c.1630C > T (p.R544x) was detected in the sporadic patient 4, which changed codon 544 from arginine (CGA) to a termination codon (TGA). Both two novel mutations were involved in exon 3 and would lead to truncated proteins without the dsRNA-binding domains and the full deaminase domain and thus compromises the enzyme activity of the ADAR1, conceivably, it must underlie the pathogenesis of DSH.
In conclusion, we have found 2 novel and 1 recurrent mutation, which will expand the database of ADAR1 gene mutations in DSH and may provide new insight into the pathogenesis of DSH. And, we also failed to find any relationship between the phenotype and genotype as the other groups have reported.
Acknowledgements
This work was funded by grants from the Natural Science Foundation of Shandong Province (ZR2011HQ003).
| 1. |
Miyamura Y, Suzuki T, Kono M, Inagaki K, Ito S, Suzuki N, et al. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am J Hum Genet 2003;73:693-9.
[Google Scholar]
|
| 2. |
Li CR, Li M, Ma HJ, Luo D, Yang LJ, Wang DG, et al. A new arginine substitution mutation of DSRAD gene in a Chinese family with dyschromatosis symmetrica hereditaria. J Dermatol Sci 2005;37:95-9.
[Google Scholar]
|
| 3. |
Song J, Zhou H, Lu RQ, Zhang LP, Sun H. The c.3463C>T mutation of the ADAR1 gene in patients with dyschromatosis symmetrical hereditaria. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2010;27:576-8.
[Google Scholar]
|
| 4. |
Li M, Yang L, Li C, Jin C, Lai M, Zhang G, et al. Mutational spectrum of the ADAR1 gene in dyschromatosis symmetrica hereditaria. Arch Dermatol Res 2010;302:469-76.
[Google Scholar]
|
Fulltext Views
1,115
PDF downloads
1,017





![[Figure - 1]](#fig_ijdvl_2012_78_6_746_102375_f1.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2012_78_6_746_102375_t3.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2012_78_6_746_102375_f2.jpg){kind=link}