Translate this page into:
Two novel TSC2 mutations in Chinese patients with tuberous sclerosis complex
2 Shandong Provincial Hospital for Skin Diseases, Provincial Academy of Medical Science; Shandong Provincial Medical Center for Dermatovenereology, Jinan, Shandong, China
3 Shandong Provincial Institute of Dermatology and Venereology; Shandong Provincial Hospital for Skin Diseases, Provincial Academy of Medical Science; Shandong Provincial Key Lab for Dermatovenereology; Shandong Provincial Medical Center for Dermatovenereology, Jinan, Shandong, China
Correspondence Address:
Furen Zhang
Shandong Provincial Institute of Dermatology and Venereology, Jingshi Road, Jinan, Shandong Province
China
| How to cite this article: You J, Liu H, Fu X, Chen M, Niu G, Tian H, Zhang F. Two novel TSC2 mutations in Chinese patients with tuberous sclerosis complex. Indian J Dermatol Venereol Leprol 2013;79:104-105 |
Sir,
Tuberous sclerosis complex (TSC, OMIM 191100) is a rare autosomal dominant disorder characterized by widespread hamartomas in several organs, including the skin, brain, kidney and eye. The cutaneous manifestations in TSC, including hypomelanotic macules, facial angiofibromas, forehead fibrous plaques, shagreen patches, ungual fibroma, and molluscum fibrosum pendulum [Figure - 1], are common and typically the first clue to the diagnosis. [1]
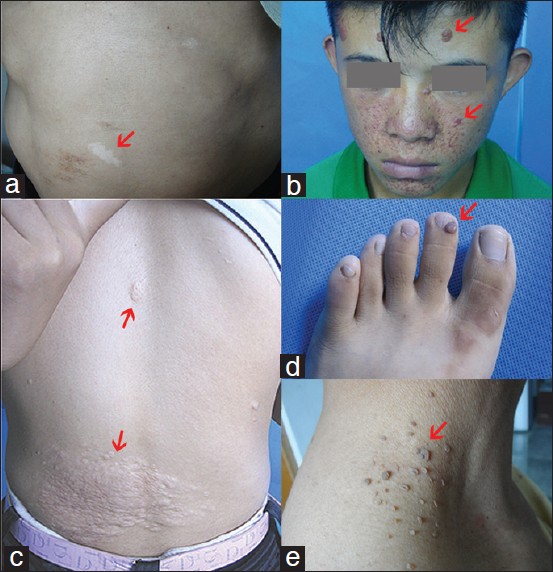 |
| Figure 1: Cutaneous features in TSC. (a) Hypomelanotic macule. (b) Facial angiofibroma. (c) Shagreen patch. (d) Ungual fibroma. (e) molluscum fibrosum pendulum |
Mutations in either the TSC1 gene on chromosome 9q34 or the TSC2 gene on chromosome 16 p13.3 cause TSC. The TSC1 and TSC2 gene products, hamartin and tuberin respectively, interact to form a protein complex. The hamartin-tuberin complex inhibits the mammalian target of rapamycin (mTOR) signaling pathway, which controls cell growth and proliferation. [2] Until now, more than 500 TSC1 and nearly 1300 TSC2 unique allelic variants have been reported (http://chromium.liacs.nl/lovd/index.php?select_db = TSC1 or db = TSC2), but there are no particular regions within the TSC1 or TSC2 gene in which mutations occur at a high rate.
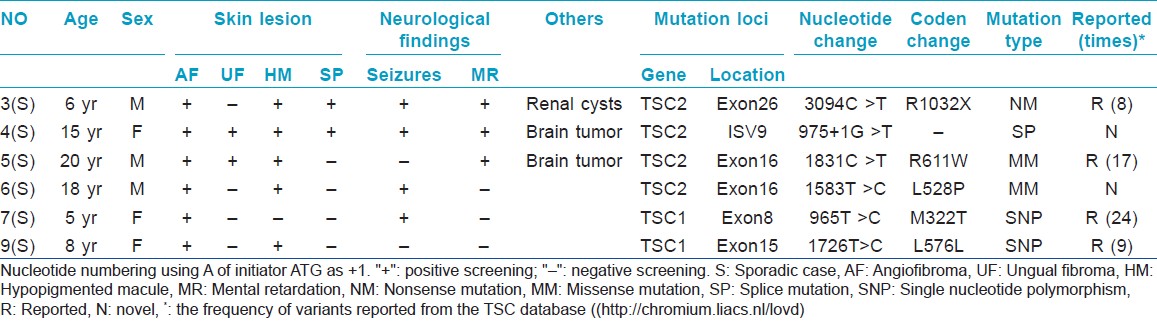
Here, we performed a genetic investigation in six sporadic Chinese TSC cases. For each case, the diagnosis was confirmed by revised diagnostic criteria for TSC and histopathological findings. After informed consent and approval of human medical and ethics committee of Shandong Provincial Institute of Dermatology and Venereology, genomic DNA was extracted from the peripheral blood of the six cases and one hundred normal healthy Chinese people. All encoding exons of TSC1 and TSC2 gene, including intron-exon boundaries, were amplified by polymerase chain reaction using the previous primers. [3] After amplification, products were purified and directly sequenced on ABI 3130XL Genetic analyzer. Two novel mutations of TSC2 were identified by comparing with the reported cDNA reference sequence (GenBank accession number: X 75621.1) and not found in one hundred controls [Table - 1].
The novel missense mutation (c.1583 T >C, p.L528P) of TSC2 has led to the change from leucine to proline, which is particularly significant for proline providing a unique conformational property which was not found in any other residues [Figure - 2]B. [4] The other novel single nucleotide substitution (c.975 + 1 G >T) occurred at the conserved 5′ splicing donor site of intron 9 [Figure - 2]a, which could result in the generation of abnormal mRNA including exon skipping, a combination of exon skipping and use of cryptic splice sites or the inclusion of intronic sequences in the mature mRNA. [5] The most likely type is the inclusion of intronic sequences in the mature mRNA which would introduce a premature stop codon (TAG) at nucleotide position c.975 + 109_111, resulting in the premature truncation of the tuberin protein which lacks two coiled-coil domains (C-C) for protein-protein interactions with hamartin, two transcription-activating domains (TAD), GAP homology, and calmodulin (CaM)-binding domains [Figure - 2]c. As these domains are functionally important and the patient exhibited no mutation except c.975 + 1 G >T, the splice site mutation was regarded as a causative mutation.
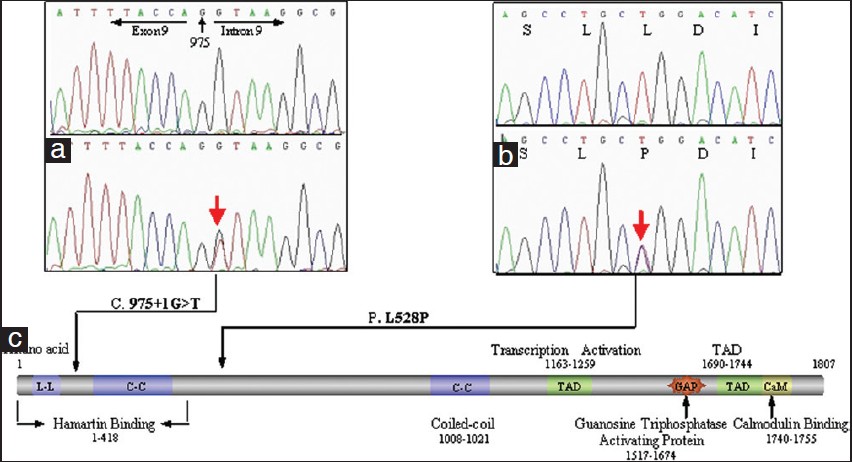 |
| Figure 2: Two novel mutations. (a) c.975 + 1 G >T in intron 9.(b) c.1583 T >C in exon 16. (c) The structure of tuberin |
Moreover, we also found two recurrent mutations (p.R611W; p.L576L) in TSC2, which were firstly detected in the Chinese Han patients, and two SNPs (p.M322T; p.L576L) in TSC1 [Table - 1]. We failed to find any relationship between the phenotype and genotype.
In summary, we have identified two novel mutations of TSC2 gene involved in Chinese TSC patients, which were predicted to be pathogenic mutations, and four recurrent variants. The cases reported supplement the available clinical and genetic data for TSC worldwide and this study can be useful for genetic counseling and prenatal diagnosis for affected families.
Acknowledgements
This work was funded by grants from the 973 Program (2011CB512105) and Project of Medical leading scholar of Shandong Province (2010). We are most grateful to all the individuals who have so willingly participated and encouraged us with these studies.
| 1. |
Rosser T, Panigrahy A, McClintock W. The diverse clinical manifestations of tuberous sclerosis complex: A review. Semin Pediatr Neurol 2006;13:27-36.
[Google Scholar]
|
| 2. |
Krymskaya VP. Tumour suppressors hamartin and tuberin: Intracellular signalling. Cell Signal 2003;15:729-39.
[Google Scholar]
|
| 3. |
Choi JE, Chae JH, Hwang YS, Kim KJ. Mutational analysis of TSC1 and TSC2 in Korean patients with tuberous sclerosis complex. Brain Dev 2006;28:440-6.
[Google Scholar]
|
| 4. |
Reiersen H, Rees AR. The hunchback and its neighbours: Proline as an environmental modulator. Trends Biochem Sci 2001;26:679-84.
[Google Scholar]
|
| 5. |
Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: Causes and consequences. Hum Genet 1992;90:41-54.
[Google Scholar]
|
Fulltext Views
1,483
PDF downloads
873





![[Figure - 1]](#fig_ijdvl_2013_79_1_104_104680_f2.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2013_79_1_104_104680_t1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2013_79_1_104_104680_f3.jpg){kind=link}