Translate this page into:
Vasculitis: Approach to diagnosis and therapy
Correspondence Address:
Arun C Inamadar
Department of Dermatology, Venereology and Leprosy, BLDEA's SBMP Medical College, Hospital and Research Centre, Bijapur - 586 103, Karnataka
India
| How to cite this article: Palit A, Inamadar AC. Vasculitis: Approach to diagnosis and therapy. Indian J Dermatol Venereol Leprol 2006;72:334-345 |


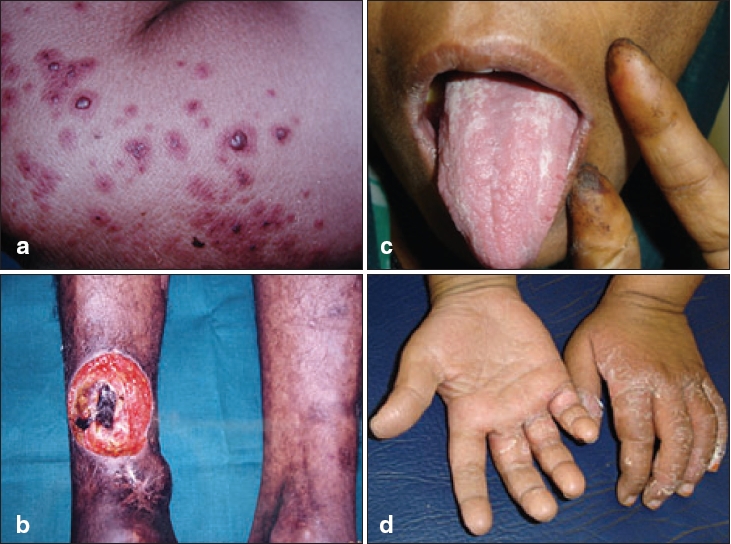 |
| (a) Palpable purpura with central hemorrhagic vesicles, (b) Vasculitic ulcer in Sjogren�s syndrome, (c) Digital tip infarct in HIV-infected patient, (d) Desquamating rash in Kawasaki disease |
|
| (a) Palpable purpura with central hemorrhagic vesicles, (b) Vasculitic ulcer in Sjogren�s syndrome, (c) Digital tip infarct in HIV-infected patient, (d) Desquamating rash in Kawasaki disease |
Introduction
Vasculitides is a group of disorders characterized by inflammation of vessel walls. The unique feature of this group is multi-organ involvement. Because of the rich vasculature, the skin is prone to be frequently affected in vasculitis. Cutaneous involvement in vasculitides may be primary or reflector of a fatal systemic disease or evidence of association with some other systemic disorder. Sometimes there may be subtle cutaneous lesions and predominant systemic involvement. These cases may present initially to other specialties, depending on the major organ of manifestation. Misdiagnosis is frequent in these situations; cases of acute abdomen, undergoing emergency exploratory laparotomy, later diagnosed as cutaneous small vessel vasculitis (CSVV), is not uncommon. Sometimes it may be necessary to assess a patient′s extent of organ involvement in several steps through clinical and laboratory examinations. Hence a systematic approach is required for diagnosis and management of these multi-system disorders.
In the following section, a diagnostic and therapeutic approach to vasculitis with cutaneous involvement relevant to the dermatologists has been discussed.
Clinical approach
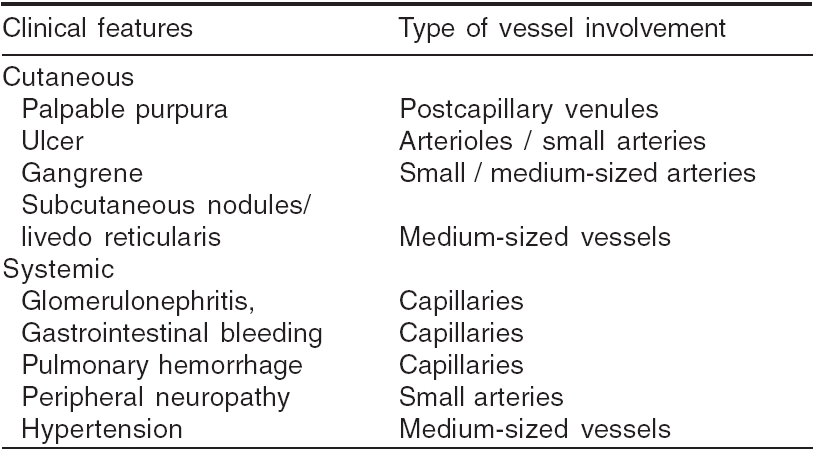
When to suspect vasculitis? The cutaneous and systemic features of vasculitides are not pathognomonic of these conditions in most of the cases. However, certain cutaneous features like palpable purpura, punched-out ulcers, livedo reticularis, subcutaneous nodules with or without certain systemic features like abdominal angina, glomerulonephritis, recurrent sinusitis, pneumonitis, peripheral neuropathy may point to a vasculitic etiology. There are certain conditions mimicking cutaneous or systemic vasculitis [Table - 1],[1] which have to be excluded.
Type of vasculitis Vasculitides represent a spectrum of disorders with overlapping cutaneous and systemic features. In some entities, cutaneous involvement predominates; in others, systemic. Some cases of CSVV, Henoch-Sch φnlein purpura (HSP), normocomplementemic urticarial vasculitis (NUV) and cutaneous PAN (c-PAN) have sole cutaneous involvement. Disorders like temporal arteritis (TA) and Takayasu′s disease (TD) develop cutaneous lesions only rarely. However, the majority of the vasculitic disorders, including hypocomplementemic urticarial vasculitis (HUV), microscopic polyangiitis (MPA), hypersensitivity vasculitis, Wegener′s granulomatosis (WG), Churg-Strauss syndrome (CSS) and classic polyarteritis nodosa (PAN) show combined features. Collagen vascular disorders, some infections and malignancies may present with vasculitic skin lesions as initial manifestation. Hence, ruling out an underlying disorder is mandatory in all cases.
Cutaneous involvement
Cutaneous features are not specific of any particular type of vasculitis.
Palpable purpura is the hallmark cutaneous sign of this group of disorders [Figure - 1]a; not observed only in cases of TA and TD.[2] In contrast to the random distribution and mucosal involvement in thrombocytopenic purpura, these lesions are common in dependent body parts, over trauma-prone sites and under tight clothing, sparing the flexures. Sometimes the purpuric lesions may show a necrotic / vesicular component or a retiform pattern. In cryoglobulinemic vasculitis (CV), skin lesions are predominantly acral[3] and in 10-30% cases cold enhancement of the purpuric lesions is observed.[4] Follicular localization of the purpuric lesions has been seen in HIV positive patients.[5] Symmetric, well-circumscribed, perifollicular papules distributed on the anterior aspect of lower limbs and scrotum is common in these patients.[5],[6]
Wheals in association with urticarial vasculitis (UV) are serpentine, symptomatic (burning sensation), tender and often show an ecchymotic component. Lesions have a predilection for the trunk and proximal extremities, are persistent (>24 hours) and leave residual hyperpigmentation. Recurrent UV may be the presenting feature in SLE (20%) and Sjogren′s syndrome (32%).[7]
Subcutaneous nodules often indicate involvement of larger vessels with systemic involvement.[3] Character and distribution of the nodule may provide clues to the underlying disorder.[2] Recurrent episodes of tender, red nodules, mainly over calves, without any systemic features is suggestive of nodular vasculitis. Erythema nodosum-like lesions may be seen in Takayasu′s arteritis.
A ′starburst′ pattern of livedo reticularis (LR), which may ulcerate, is the predominant cutaneous feature in c-PAN.[1] Livedo reticularis is rare in CV and CSS and unusual in WG.[1] A combination of LR and nodules is indicative of larger vessel and systemic involvement[3] and LR preceding and following the subcutaneous nodules is observed in SLE and classic PAN respectively.[2] Cutaneous small vessel vasculitis may occasionally present with livedoid pattern of the vasculature.
Ulceration is of common occurrence, especially with involvement of medium to large-sized vessels. Necrotic papules or nodules may form ulceration in WG; however, pyoderma gangrenosum (PG) like ulcers, which lack the characteristic rolled-out border of idiopathic PG, are pathognomonic of this condition.[1] Ulcers in the peri-anal location are also common in WG. Ischemic ulcers are seen with arterial involvement in their distribution like scalp ulceration in temporal arteritis.[1] Punched out ulcers are seen in vasculitis with underlying HIV infection or collagen vascular disorders like SLE and Sjogren′s syndrome [Figure - 1]b.
Raynaud′s phenomenon and peripheral cyanosis are common in CV (20-50%).[4] Pale, cold extremities are seen in giant cell arteritis in association with asymmetric peripheral pulses. Digital gangrene is common in conditions with involvement of the larger vessels like classic PAN. HIV associated vasculitis may manifest as digital tip infarction [Figure - 1]c.
A nondescript maculopapular rash which subsides with periungual and perineal desquamation is characteristic of Kawasaki disease [Figure - 1]d. HIV-infected patients with vasculitis may present with a Kawasaki-like syndrome.[8] An asymptomatic, permanent erythema of the fingers (red finger syndrome), may be the presenting feature of HIV-associated necrotizing vasculitis (2.4%), seen more commonly among intravenous drug abusers.[9]
Mucosal involvement
Persistent oral and nasal mucosal ulcers are common in WG. Hyperplastic gingivitis with petechiae (strawberry gingival hyperplasia), is a pathognomonic feature of WG.[2]
Nasal polyps, persistent for years are seen in CSS. Recurrent pansinusitis of long duration may be the presenting feature of WG before other organ involvements are evident. Septal perforation and saddle-nose deformity, though rare, is strongly suggestive of WG.
Cheilitis (dry, fissured lips) and strawberry tongue in association with a desquamating rash is suggestive of Kawasaki disease. A swollen, cyanotic, cold, tender tongue, which later becomes atrophic, may result due to ischemia in TA.
Systemic involvement
When encountered with a patient having cutaneous features of vasculitis, the initial attempt should be to assess the extent of systemic involvement. Though systemic manifestations are common in primary systemic vasculitides, some primary cutaneous disorders may also have associated organ involvement. Organ involvement may be present even in the absence of overt clinical features. The extent and severity of systemic involvement determines the prognosis of the disease. Moreover, the diagnostic and therapeutic approach to the patient is dependent on this.
Gastrointestinal symptoms like abdominal angina, hematemesis, melena, bloody diarrhea and intestinal obstruction are more common with small vessel vasculitis like CSVV, HSP and urticarial vasculitis. Presentation with an acute abdomen is quite common (50-85%) in HSP and about one-third of these cases present with gastrointestinal hemorrhage.[10] Lanzkowsky et al[11] have reported that 14% patients in their series had gastrointestinal symptoms preceding cutaneous lesions and diagnosis may become difficult in this situation. The interval between abdominal symptoms and appearance of skin lesions in such cases may be as long as 2 to 24 weeks.[12],[13]
Renal involvement is variable; glomerulonephritis is common in different types of cutaneous vasculitis. Membrano-proliferative glomerulonephritis (MPGN) is typical of CV.[1] Marked renal involvement is a feature of classic PAN; reno-vascular hypertension and renal failure are important diagnostic clues to this disorder. A pulmonary-renal syndrome (crescentic, necrotizing glomerulonephritis, pulmonary hemorrhage + circulating antineutrophil cytoplasmic antibody [ANCA]) is seen in WG, CSS and most commonly in MPA.[14] Renal involvement in CSS is comparatively uncommon and less severe than WG and MPA.
Asthma is the central feature of CSS[15] which is not usual in WG. Almost always it precedes other systemic manifestations[15] and unlike atopic individuals, there is a late onset, at a mean age of 35 years.[1] Lungs and spleen are usually spared in classic PAN.[1] A granulomatous myocarditis is characteristic of CSS and not found in other disorders in this group.[16]
Arthralgia / arthritis are frequently associated with CSVV, HSP, CV, UV, MPA and PAN. Peripheral neuropathy and mononeuritis multiplex involving mainly lower limbs is seen at highest frequency in CSS but is also common in CV, c-PAN, classic PAN and vasculitis associated with rheumatoid arthritis.[17],[18] Orchitis is a feature of classic PAN, more frequently seen in hepatitis B infection associated cases.[19]
Patients with HUV have systemic manifestations like arthritis, asthma and gastrointestinal symptoms more commonly than patients with the normocomplementemic disease.[1]
There is an overlap of clinical features (persistent urticarial wheals, pleuritis and glomerulonephritis) between hypocomplementemic urticarial vasculitis syndrome (HUVS) and SLE.[1],[20] Angioedema, ocular inflammation (iritis, episcleritis, uveitis) and obstructive pulmonary disease are common in HUVS. Renal involvement is severe in SLE but mild in HUVS.
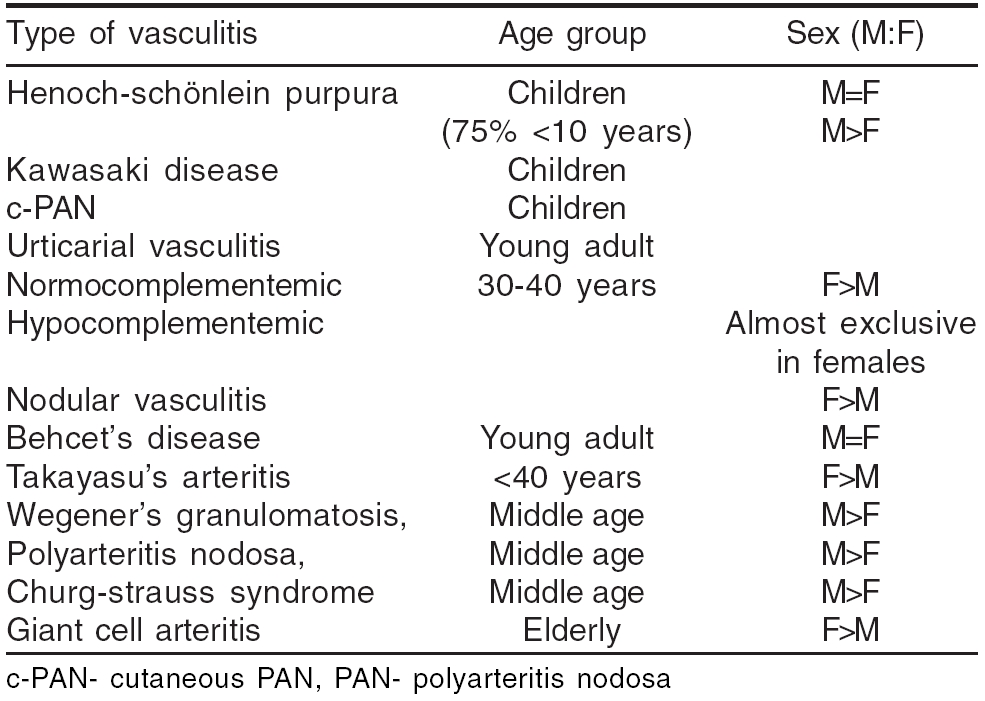
Age and sex predilection has been observed in some of the vasculitic disorders [Table - 2].
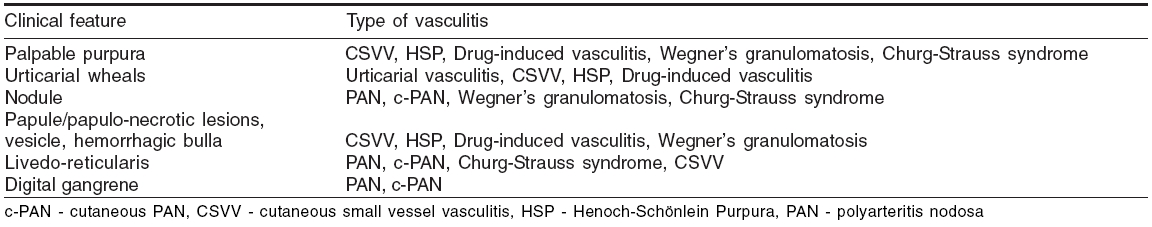
Clinical presentations indicative of the type of the vasculitis have been listed in [Table - 3].[21] Approximate idea of the nature of vessel involvement is possible from some of the presenting features [Table - 4].[21]
Looking for treatable etiology Cutaneous vasculitis is idiopathic in 50% cases.[22] However, evaluation of a patient with suspected vasculitis includes intensive search to find out any treatable underlying cause. Infections (15-20%), inflammatory disorders (15-20%), drugs (10-15%) and malignancies (2-5%) are known precipitating factors for vasculitis.[23],[24]
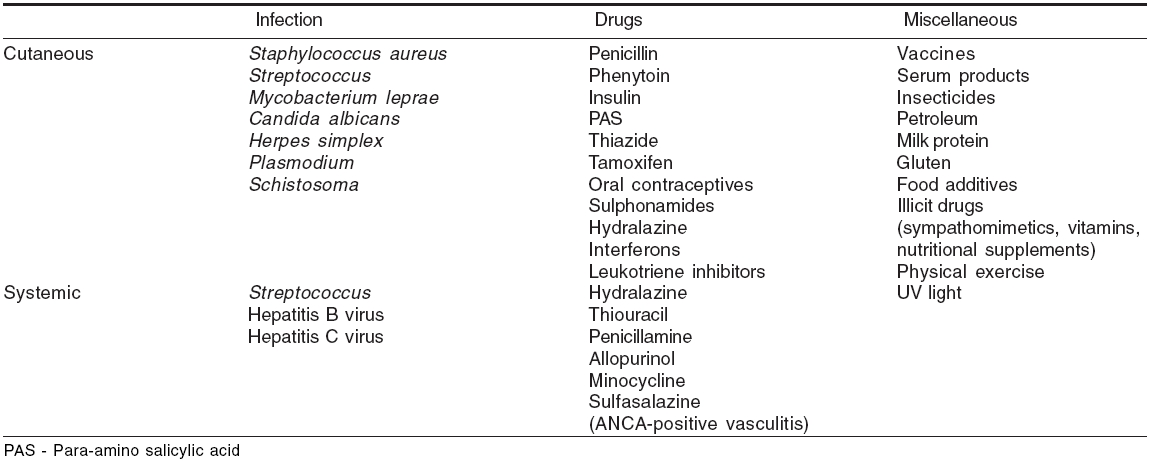
Approximately 5-7% cases of classic PAN are associated with hepatitis B virus infection.[19],[25] As observed by some authors, approximately 20% patients with cutaneous vasculitis show a positive serology for HCV.[26],[27] CV has been associated with hepatitis C virus infection (75%), autoimmune diseases (24%) and lymphoproliferative disorders (6%).[27] Cutaneous PAN has been associated with various types of infections ( Streptococcus , Parvovirus B-19, HIV, hepatitis B), inflammatory bowel disease and inferior vena cava thrombosis.[1] The classical example of malignancy-associated vasculitis is occurrence of PAN with hairy cell leukemia.[28] In HIV-infected patients, vasculitis may be precipitated by cryoglobulinemia, drugs and infections like cytomegalovirus / Parvovirus B19 / HIV per se.[1],[29]
Infections and other agents which may be overlooked as trigger factors for vasculitis have been listed in [Table - 5].[6] Some disorders presenting with vasculitic skin lesions are presented in [Table - 6].[6]
Diagnostic approach
Thorough history and physical examination of patients help in initial clinical diagnosis. Criteria for diagnosis of both cutaneous and systemic vasculitides proposed by American College of Rheumatology (ACR, 1990)[1] may be used for categorization to the type of vasculitis. Recently, a clinical classification system for vasculitis has been proposed by Sunderk φtter et al[30] focusing on the types of vasculitis commonly encountered by dermatologists. Laboratory tests are required for confirmation of the clinical diagnosis, screening for underlying organ involvement and occasionally, to assess the risk factors, prognosis or response to treatment.
Confirmation of clinical diagnosis
Role of histopathological examination Vasculitis is a clinico-pathological diagnosis and microscopic evidence of vascular inflammation is confirmatory. Skin, being the most accessible organ, is most frequently sampled for this purpose.
A deep punch / incision biopsy is taken from a lesion of appropriate stage. Optimum timing for skin biopsy for this purpose is within 18-36 hours of the onset of the lesions.[31] In case of CSVV, lesions from the uppermost part of the limb (early lesion) are chosen. Deeper, elliptical, incision biopsy is required when lesions are suggestive of larger vessel involvement, e.g. presence of nodules.[3] Upright position makes the nodular lesions more prominent; hence the patient can be asked to stand up for a few moments before selecting a nodule for biopsy.[23]
Segmental inflammation of the vessel wall, fibrinoid necrosis, endothelial swelling and leucocytoclasia are the major features of CSVV. Mostly small-vessel involvement with lymphocytic infiltrate, little leukocytoclasia and some degree of tissue eosinophilia are suggestive of drug-induced vasculitis. Leucocytoclasia is also a histopathological finding in both types of urticarial vasculitis, but these two can be distinguished by the presence of large numbers of interstitial neutrophils in case of HUV and eosinophils in NUV.[32] Lesions younger or older than the specified period may show a predominant lymphocytic infiltrate and findings like leucocytoclasia and fibrinoid necrosis may be absent.[6]
Though sensitive, skin biopsy may not be specific or sufficient to establish diagnosis of PAN, WG and CSS. Leukocytoclasia, a feature common to most of the vasculitides, is a frequent histopathological finding in these cases rather than specific features like granulomatous vasculitis.[31] In these disorders, as and when appropriate, sural nerve biopsy (in presence of abnormal nerve conduction velocity), muscle biopsy, open lung biopsy, percutaneous kidney biopsy (in presence of persistent, abnormal urinary sediment) or testicular biopsy may be indicated. Nasal tissue biopsy, though easier to perform, has a far lower potential as diagnostic help than lung biopsy in cases with WG.[33]
Role of immunofluorescence study For direct immunofluorescence (DIF) study, the skin sampling should be done from a fresh, noninfarcted, most proximal lesion (< 6 hours duration).[31] Biopsy from a dependent area, e.g., lower leg may demonstrate nonspecific fluorescence resulting from hydrostatic extravasation of immune complex.[3] DIF is not conclusive if the biopsy is taken beyond 24 hours, as the immunoreactants are likely to be phagocytized by this time.
The most common immune deposits in blood vessels are IgM, C3 and fibrin.[3] Though not specific, IgA deposit within the vessel wall distinguishes HSP from other cutaneous small vessel vasculitis. Demonstration of this feature increases the sensitivity of diagnosis in patients with HSP who have insufficient clinical criteria.[23] Vascular and basement membrane zone fluorescence, predominantly with C3 observed in HUV and can help in differentiation from NUV.[3] Granular IgG deposition with occasional IgM, IgA and C1, C3 at the basement membrane zone is observed in 80% cases of SLE and frequently in HUV.[3],[23]
Histopathological study of skin biopsy is confirmatory, while DIF study is additive to this and helps to categorize the vasculitis.
Screening for underlying organ involvement and other laboratory tests This involves a battery of laboratory tests like routine hemogram, ESR, urinalysis, throat swab for culture and sensitivity test, ASLO titer and C-reactive protein (CRP). Leukopenia or thrombocytopenia is almost never seen in primary vasculitic disorders and the presence of these may be indicative of underlying conditions like SLE, malignancy or it may be drug-induced.[33] Marked eosinophilia (>1000/mL) is a feature of CSS and helps in differentiation from WG.[33] Urinalysis may reveal proteinuria, hematuria or presence of casts indicative of renal involvement. An abnormal liver function test may be indicative of underlying viral hepatitis (B or C).
Estimation of serum complement levels is helpful as these are often lowered in CSVV, CV (90%), UV (18%) and vasculitis associated with collagen vascular disorders.[1] Complement levels are usually normal in HSP. Anti-C1q antibodies are present more commonly in patients with HUVS (100%) than SLE (30%) and may help in differentiating between these two conditions.[34] Estimation of cryoglobulin levels is an important parameter, as in addition to CV, it may be indicative of associated hepatitis C infection.[1],[35]
Role of ANCA Estimation of ANCA in patients with clinical features suggestive of WG / CSS / MPA is recommended as this may postpone the immediate need of invasive lungs / kidney biopsy and help in early diagnosis.[1] However, this test is not absolutely sensitive or specific.[1] Hence confirmation of diagnosis requires a biopsy of the involved organ. C-ANCA is reasonably specific for WG (75-80%) and MPA (25-30%).[1] This is detected in only 10-15% cases of CSS. P-ANCA is less specific, detected in MPA (50-60%), CSS (55-60%), WG (10-15%) and some other conditions like drug-induced vasculitis, rheumatoid arthritis (30-70%), SLE (20-30%), ulcerative colitis (50-70%), Crohn′s disease (20-40%) and different hepatic disorders.[1] If C-ANCA and P-ANCA coexist in a patient, drug-induced vasculitis should be suspected.[36]
Hence the role of ANCA is in screening for these disorders and should be asked for only when highly suggestive clinical features like pulmonary hemorrhage, recurrent sinusitis orbital mass or glomerulonephritis are present.[1] Estimation of ANCA level is also helpful in disease monitoring. ANCA is rarely (< 5%) positive in CV[37] and almost always negative in Takayasu′s disease, temporal arteritis and Kawasaki disease.[38]
Serology for HIV, hepatitis B and C viruses, immunological tests like rheumatoid factor (RA factor), antinuclear antibody (ANA) and anti ds-DNA are helpful screening tests for associated disorders. However, ANA may be positive in patients with UV (low titer, 30-50%), HUVS and CV (20%).[1] Anti ds-DNA positivity is also seen in HUVS and RA factor positivity in patients with CV (>70%).[1]
Endoscopy may be of diagnostic help in cases of HSP with gastrointestinal symptoms as presenting features. Coin-like petechiae, hemorrhagic erosions, skip hyperemia and ecchymosis are the lesions suggestive of vasculitis.[10] In patients with palpable purpura and poorly localized colicky abdominal pain, CT scan / ultrasonography may be helpful to assess the extent of gastrointestinal involvement. The hallmark findings in CT scan are focal areas of bowel thickening, mesenteric edema, nonspecific lymphadenopathy and vascular engorgement.[39] Intussusception and ascites are easily detectable by ultrasonography.[10]
Identifying risk factors From the dermatologists′ point of view, the majority of the patients with skin-limited disorder have a good prognosis. However, a disorder which appears to be limited to the skin, may have asymptomatic / minimally symptomatic widespread systemic involvement or may subsequently develop so. Hence identification of the risk factors in individual cases may modify the therapeutic approach.
In CSVV and HSP, predictors of renal involvement include spread of the palpable purpura above the waist, presence of fever and elevated ESR.[40] Adult patients with HSP suffer from more severe nephritis. In 8-10% cases of CSVV, there are recurrent crops of palpable purpura indicating chronicity of the disease.[3] As observed by Sais et al .,[26] factors predicting chronicity of cutaneous lesions were, presence of arthralgia and cryoglobulinemia and absence of fever. Though nonspecific, raised ESR and CRP are markers of disease activity in most cases of vasculitis.
In patients with classic PAN, a five factor score (FFS) is in use to detect the risk of mortality and this includes serum creatinine > 1.58 mg/dl, proteinuria > 1g / day, gastrointestinal involvement (bleeding/ perforation/ infarction / pancreatitis), CNS involvement and cardiomyopathy.[41] Activity of skin lesions in patients with WG parallels that of systemic disease.[38] Presence of pulmonary hemorrhage (10%) is a bad prognostic factor in patients with pulmonary-renal syndrome with a mortality as high as 50%.[42] Granulomatous myocarditis is a leading cause of death in patients with CSS.[16] A significant rise in C-ANCA titer in patients with WG, carry a 50% risk of relapse of the disease within six months.[1] ANCA-negative cases of WG have limited disease with a better prognosis.[43]
In CV, serum cryoglobulin levels do not correlate with the disease activity, but C3 level fluctuates with the disease course.[1]
Cutaneous vasculitic lesions in SLE are indicative of flare-up of the disease organ involvement like the central nervous system and others and carry a poor prognosis.[44] Punched-out ulcers in SLE and Sjogren′s syndrome warrant aggressive therapy as these are indicative of systemic vasculitis.[1]
Therapeutic approach
General measures In the active stage of the disease, the basic instructions include avoiding stress, bed rest, elevation of the foot end and keeping extremities warm. Patients with major pulmonary involvement, like in UV and CSS, are advised to avoid smoking. Antihistamines and NSAIDs reduce symptoms like pruritus and joint pain. Specific therapies are aimed at reducing acute symptoms and preventing complications.
Specific therapy In cases with underlying disorder, the vasculitic lesions usually resolve with control of the infection, withdrawal of the causative drug or removal of a tumor. Choice of therapy in vasculitic disorders is experience-based.[23] In many of the cases, a chronic course presents a difficult situation; hence an effective but least toxic therapeutic regimen is preferred.
Role of neutrophil-chemotaxis inhibitors Dapsone (50-200 mg/day in divided doses) and colchicine (0.6-1.8 mg/day in divided doses) are the most effective initial agents for treatment of CSVV and similar disorders in the absence of evidence of systemic involvement.[35] Either of these two drugs can be started in an acute episode of the disease and response is observed within two weeks. However, as experienced by several authors, a subset of patients does not respond to either colchicine or dapsone and may require alternative treatment.[34] Recurrent cases may also need these alternative therapies. In cases of UV, dapsone/colchicine may be combined with indomethacin or hydroxychloroquine.[45],[46]
Role of antimalarials Hydroxychloroquine (200-400 mg/day) has been used effectively in HUV but not in other small vessel vasculitides.[3]
Role of corticosteroid In small vessel vasculitis not responding to dapsone and/or colchicine, low-dose corticosteroid (< 10 mg on alternate days) may be helpful in alleviating symptoms.[35] However, long-term corticosteroid monotherapy is not recommended because of related side-effects.
Indications for systemic steroid as first-line therapy include severe ulcerative / necrotic cutaneous lesion, gastrointestinal bleeding, acute glomerulonephritis and peripheral neuropathy with impending palsy. These situations necessitate high-dose corticosteroid (60-80 mg/day), tapered over several weeks.
In children with HSP, administration of systemic steroid at presentation, may prevent risk of developing nephritis and in combination with immunomodulatory drugs (azathioprine/cyclosporine), may improve the outcome of existing nephritis.[47] Patients with c-PAN often require high-dose corticosteroid, tapered slowly, in combination with aspirin/NSAID.[48] In classic PAN, disease control can be achieved with corticosteroids with/without cyclophosphamide. However, in hepatitis B virus infection associated cases, such therapy should be short-term to prevent the risk of viral replication.[25] Short-term systemic steroid (0.5-1.5 mg/kg/day) may also be used to treat renal and CNS manifestations of HCV-associated CV.[1]
High-dose corticosteroid (1 mg/kg/day) is the initial therapeutic option for WG, MPA and CSS. Intravenous ′pulse′ therapy (methylprednisolone 1 g / day ´ 3 days) can be administered to patients with life-threatening organ involvement.[21] Systemic corticosteroids are relatively contraindicated in Kawasaki disease to avoid the risk of coronary aneurysm formation.[21]
Role of immunomodulators Weekly dosage of methotrexate (up to 25 mg / week), azathioprine (2 mg/kg/day), cyclophosphamide or cyclosporine constitutes the alternative therapeutic regimen in cases of CSVV not responding to neutrophil chemotaxis inhibitors.[35] Addition of immunomodulators to corticosteroid while treating cases of WG, MPA and CSS with significant systemic involvement induces remission and increases patient survival rate, though with increased risk of toxicity.[1] Monthly pulse of intravenous cyclophosphamide can be used in WG to avert the risk of toxicity.[1] Azathioprine prevents recurrence in cases of CSVV either used alone or in combination with low-dose corticosteroid.[3]
Role of antiviral drugs In hepatitis C virus-associated cases of CV, interferon a (IFN a) is the preferred drug.[1],[49] The treatment schedule consists of 3 million IU of IFN a, thrice weekly, for a total duration of 12 to 18 months.[1] Significant improvement (60-80%) regarding cutaneous, renal and joint manifestations has been observed with this therapy along with decrease in cryoglobulin level.[50] However, systemic features respond slowly and incompletely with a relapse rate as high as 90%. Ribavirin, with / without IFN a, may be used for treatment or prevention of relapse. HBV-associated PAN may need antiviral treatment (IFN a2 ± vidarabine ± lamivudine) in combination with plasma exchange.[1]
Role of Intravenous immunoglobulin G (IVIgG) The treatment of choice for Kawasaki disease is IVIgG (2 g/kg single dose) in combination with high-dose aspirin.[33] Early administration of IVIgG in these patients prevents future risk of aneurysm formation.[33] It also improves cutaneous as well as systemic involvement (gastrointestinal and renal) in patients with HSP.[51] There are reports of successful treatment of c-PAN with IVIg (2 g / kg over 2-5 days), but relapse may occur after several months.[1],[52]
Newer treatment modalities Mycophenolate mofetil has been used successfully in WG and MPA.[3] This is also effective in maintaining remission in HUV.[53] Infliximab has been used in steroid nonresponsive necrotizing CSVV.[54]
Miscellaneous Penicillin is often used as treatment or prophylaxis in childhood cases of c-PAN associated with streptococcal infection.[1] Isolated upper respiratory tract relapses of WG are often precipitated by infection with Staphylococcus aureus. Some workers have used co-trimoxazole in association with regular therapeutic regimen and found it effective in maintaining remission in this condition.[1] Prostaglandin I 2 (50 mg / day IV) and nifedipine (60 mg / day) has been tried in cases of c-PAN associated with digital gangrene.[1]
In all cases of vasculitis, the attempt should be to reach a definite diagnosis rather than using broad, nonspecific terminologies. This needs a comprehensive approach towards understanding of the disease. Determining prognosis and deciding therapeutic strategy in these cases depend mostly on the extent of organ involvement. The morbidity and mortality associated with these disorders can be significantly reduced if recognized and treated early. Since some form of cutaneous involvement is a universal finding in all types of vasculitis, dermatologists play a crucial role in diagnosing vasculitic disorders, predicting the extent of underlying systemic involvement and prognosis.
| 1. |
Fiorentino DF. Cutaneous vasculitis. J Am Acad Dermatol 2003;48:311-40.
[Google Scholar]
|
| 2. |
Piette WW. Primary systemic vasculitis. In: Sonthiemer RD, Provost TT, editors. Cutaneous manifestations of rheumatic disease. Baltimore: Williams and Wilkins; 1996. p. 177-232.
[Google Scholar]
|
| 3. |
Russell JP, Gibson LE. Primary Cutaneous small vessel vasculitis: Approach to diagnosis and treatment. Int J Dermatol 2006;45: 3-13.
[Google Scholar]
|
| 4. |
Brouet JC, Clauvel JP, Danon F, Klein M, Seligmann M. Biologic and clinical significance of cryoglobulins. A report of 86 cases. Am J Med 1974;57:775-88.
[Google Scholar]
|
| 5. |
Weimer CE, Jr, Sahn EE. Follicular accentuation of leucocytoclastic vasculitis in an HIV-seropositive man: Report of a case and review of the literature. J Am Acad Dermatol 1991;24:898-902.
[Google Scholar]
|
| 6. |
Lotti T, Ghersetich I, Comacchi C, Jorizzo JL. Cutaneous small-vessel vasculitis. J Am Acad Dermatol 1998;39:667-87.
[Google Scholar]
|
| 7. |
Black AK. Urticarial vasculitis. Clin Dermatol 1999;17:565-9.
[Google Scholar]
|
| 8. |
Johnson RM, Little JR, Storch GA. Kawasaki-like syndromes associated with human immunodeficiency virus infection. Clin Infect Dis 2001;32:1628-34
[Google Scholar]
|
| 9. |
Abajo P, Porras-Luque JI, Buezo GF, Fraga J, Dauden E. Red finger syndrome associated with necrotizing vasculitis in an HIV-infected patient with hepatitis B. Br J Dermatol 1998;139:154-5.
[Google Scholar]
|
| 10. |
Chen MJ, Wang TE, Chang WH, Tsai SJ, Liao WS. Endoscopic findings in a patient with Henoch-Sch φnlein purpura. World J Gastroenterol 2005;11:2354-6.
[Google Scholar]
|
| 11. |
Lanzkowsky S, Lanzkowsky L, Lanzkowsky P. Henoch-Schoenlein purpura. Pediatr Rev 1992;13:130-7.
[Google Scholar]
|
| 12. |
Van der Boon F, Groeneweg M. Acute abdominal pain as the first sign of Henoch-Sch φnlein purpura; A hidden diagnosis in the absence of purpura. Ned Tijdschr Geneeskd 2005;149:2522-6. (Abstract).
[Google Scholar]
|
| 13. |
Nathan K, Gunasekaran TS, Berman JH. Recurrent gastrointestinal Henoch-Sch φnlein purpura. J Clin Gastroenterol 1999;29:86-9.
[Google Scholar]
|
| 14. |
Niles JL, Bottinger EP, Saurina GR, Kelly KJ, Pan G, Collins AB, et al . The syndrome of lung hemorrhage and nephritis is usually an ANCA-associated condition. Arch Intern Med 1996;156: 440-5.
[Google Scholar]
|
| 15. |
Guillevin L, Pragnoux C, Mouthon L. Churg Strauss syndrome. Semin Respir Crit Care Med 2004;25:535-45.
[Google Scholar]
|
| 16. |
Lhote P, Guillevin L. Polyarteritis nodosa, microscopic polyangitis and Churg-Strauss syndrome: Clinical aspects and treatment. Rheum Dis Clin North Am 1995;21:911-47.
[Google Scholar]
|
| 17. |
Guillevin L, Cohen P, Gayraud M, Lhote F, Jarrousse B, Casassus P. Churg-Strauss syndrome. Clinical study and long-term follow-up of 96 patients. Medicine (Baltimore) 1999;78:26-37.
[Google Scholar]
|
| 18. |
Stone JH, Catabrese LH, Hoffman GS, Pusey CD, Hunder GG, Hellman DB. Vasculitis. A collection of pearls and myths. Rheum Dis Clin North Am 2001;27:677-728.
[Google Scholar]
|
| 19. |
Lhote F, Cohen P, Guillevin L. Polyarteritis nodosa, microscopic polyangiitis and Churg-Strauss syndrome. Lupus 1998;7: 238-58.
[Google Scholar]
|
| 20. |
Wisnieski JJ, Baer AN, Christensen J, Cupps TR, Flagg DN, Jones JV, et al . Hypocomplementemic urticarial vasculitis syndrome. Clinical and serologic findings in 18 patients. Medicine 1995;74:24-41.
[Google Scholar]
|
| 21. |
Roane DW, Griger DR. An approach to diagnosis and initial management of systemic vasculitis. Am Fam Physician 1999;60:1421-30.
[Google Scholar]
|
| 22. |
Barham KL, Jorizzo JL, Grattan B, Cox NH. Vasculitis and neutrophilic vascular reactions. In : Burns T, Breathnach S, Cox NH, Griffiths C, editors. Rook's Text Book of Dermatology, 7th ed. Blackwell Science: Oxford; 2004. p.49.1-46.
[Google Scholar]
|
| 23. |
Gibson LE. Cutaneous vasculitis update. Dermatol Clin 2001;19:603-15.
[Google Scholar]
|
| 24. |
Blanco R, Martinez-Taboada VM, Rodriguez-Valverde V, Garcia-Fuentes M. Cutaneous vasculitis in children and adults. Associated diseases and etiologic factors in 303 patients. Medicine (Baltimore) 1998;77:403-18.
[Google Scholar]
|
| 25. |
Guillevin L, Lhote F, Cohen P, Sauvaget F, Jarrousse B, Lortholary O, et al . Polyarteritis nodosa related to hepatitis B virus. A prospective study with long-term observation of 41 patients. Medicine (Baltimore) 1995;74:238-53.
[Google Scholar]
|
| 26. |
Sais G, Vidaller A, Jucgla A, Servitje O, Condom E, Peyri J. Prognostic factors in leukocytoclastic vasculitis: A clinicopathologic study of 160 patients. Arch Dermatol 1998;134:309-15.
[Google Scholar]
|
| 27. |
Trejo O, Ramos-Casals M, Garcta-Carrasco M, Yague J, Jimenez S, de la Red G, et al . Cryoglobulinemia: Study of etiologic factors and clinical and immunologic features in 443 patients from a single center. Medicine 2001;80:252-62.
[Google Scholar]
|
| 28. |
Greer JM, Longley S, Edwards NL, Elfenbein GJ, Panush RS. Vasculitis associated with malignancy. Experience with 13 patients and literature review. Medicine (Baltimore) 1988;67:220-30.
[Google Scholar]
|
| 29. |
Johnson RA. Cutaneous manifestations of Human immunodeficiency virus disease. In : Freedberg IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz SI, editors. Fitzpatrick's Dermatology in General Medicine, 6th ed. McGraw-Hill: New York; 2003. p. 2138-50.
[Google Scholar]
|
| 30. |
Sunderk φtter C, Sindrilaru A. Clinical classification of vasculitis. Eur J Dermatol 2005;16:114-24.
[Google Scholar]
|
| 31. |
Sams WM Jr. Vasculitis. In : Lebwohl M, editor. Difficult diagnoses in dermatology. Churchill Livingstone: New York; 1988. p. 1-9.
[Google Scholar]
|
| 32. |
Davis MD, Daoud MS, Kirby B, Gibson LE, Rogers RS 3rd. Clinicopathologic correlation of hypocomplementemic and normocomplementemic urticarial vasculitis. J Am Acad Dermatol 1998;38:899-905.
[Google Scholar]
|
| 33. |
Mandell BF, Hoffman GS. Systemic necrotizing arteritis. In : Freedberg IM, Eisen AZ, Wolff K, Austen KF, Goldsmith LA, Katz SI, editors. Fitzpatrick's Dermatology in General Medicine, 6thed. McGraw-Hill: New York; 2003. p. 1718-26.
[Google Scholar]
|
| 34. |
Wisnieski JJ, Jones SM. IgG autoantibody to the collagen-like region of C1q in hypocomplementemic urticarial vasculitis syndrome, systemic lupus erythematosus and 6 other musculoskeletal or rheumatic diseases. J Rheumatol 1992;19:884-8.
[Google Scholar]
|
| 35. |
Callen JP. Cutaneous vasculitis. What have we learned in the past 20 years? Arch Dermatol 1998;134:355-7.
[Google Scholar]
|
| 36. |
Dolman KM, Gans RO, Vervaat TJ, Zevenbergen G, Maingay D, Nikkels RE, et al . Vasculitis and antineutrophil cytoplasmic autoantibodies associated with propylthiouracil therapy. Lancet 1993;342:651-2.
[Google Scholar]
|
| 37. |
Lamprecht P, Gause A, Gross WL. Cryoglobulinemic vasculitis. Arthritis Rheum 1999;42:2507-16.
[Google Scholar]
|
| 38. |
Burrows NP, Lockwood CM. Antineutrophil cytoplasmic antibodies and their relevance to the dermatologist. Br J Dermatol 1995;132:173-81.
[Google Scholar]
|
| 39. |
Jeong YK, Ha HK, Yoon CH, Gong G, Kim PN, Lee NG, et al . Gastrointestinal involvement in Henoch-Sch φnlein syndrome: CT findings. Am J Roentgenol 1997;168:965-8.
[Google Scholar]
|
| 40. |
Tancrede-Bohin E, Ochonisky S, Vignon-Pennamen MD, Flageul B, Morel P, Rybojad M. Sch φnlein-Henoch purpura in adult patients. Predictive factors for IgA glomerulonephritis in a retrospective study of 57 cases. Arch Dermatol 1997;133: 438-42.
[Google Scholar]
|
| 41. |
Guillevin L, Lhote F, Gayraud M, Cohen P, Jarrousse B, Lortholary O, et al . Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine 1996;75:17-26.
[Google Scholar]
|
| 42. |
Falk RJ, Jennette JC. ANCA small-vessel vasculitis. J Am Soc Nephrol 1997;8:314-22.
[Google Scholar]
|
| 43. |
Homer RJ. Antineutrophil cytoplasmic antibodies as markers for systemic autoimmune disease. Clin Chest Med 1998;19:627-39,viii.
[Google Scholar]
|
| 44. |
Hautmann G, Campanile G, Lotti TM. The many faces of cutaneous vasculitis. Clin Dermatol 1999;17:515-31.
[Google Scholar]
|
| 45. |
Mehregan DR, Hall MJ, Gibson LE. Urticarial vasculitis: A histopathologic and clinical review of 72 cases. J Am Acad Dermatol 1992;26:441-8.
[Google Scholar]
|
| 46. |
Lopez LR, Davis KC, Kohler PF, Schocket AL. The hypcomplementemic urticarial vasculitis syndrome: Therapeutic response to hydroxychloroquine. J Allergy Clin Immunol 1984;73:600-3.
[Google Scholar]
|
| 47. |
Mollica F, Li Volti S, Garozzo R, Russo G. Effectiveness of early prednisone treatment in preventing the development of nephropathy in anaphylactoid purpura. Eur J Pediatr 1992;151:140-4.
[Google Scholar]
|
| 48. |
Sheth AP, Olson JC, Esterly NB. Cutaneous polyarteritis nodosa of childhood. J Am Acad Dermatol 1994;31:561-6.
[Google Scholar]
|
| 49. |
Cresta P, Musset L, Cacoub P, Frangeul L, Vitour D, Poynard T, et al . Response to interferon alpha treatment and disappearance of cryoglobulinaemia in patients infected by hepatitis C virus. Gut 1999;45:122-8.
[Google Scholar]
|
| 50. |
Misiani R, Bellavita P, Fenili D, Vicari O, Marchesi D, Sironi PL, et al . Interferon alpha-2a therapy in cryoglobulinemia associated with hepatitis C virus. N Eng J Med 1994;330:751-6.
[Google Scholar]
|
| 51. |
Lamireau T, Rebouissoux L, Hehunstre JP. Intravenous immunoglobulin therapy for severe digestive manifestations of Henoch-Sch φnlein purpura. Acta Pediatr 2001;90:1081-2.
[Google Scholar]
|
| 52. |
Gedalia A, Sorensen R. Intravenous immunoglobulin in childhood cutaneous polyarteritis nodosa. Clin Exp Rheumatol 1998;16:767.
[Google Scholar]
|
| 53. |
Worm M, Sterry W, Kolde G. Mycophenolate mofetil is effective for maintenance therapy of hypocomplementaemic urticarial vasculitis. Br J Dermaol 2000;143:1324.
[Google Scholar]
|
| 54. |
Mang R, Ruzicka T, Stege H. Therapy for severe necrotizing vasculitis with infliximab. J Am Acad Dermatol 2004;51: 321-2.
[Google Scholar]
|
Fulltext Views
13,591
PDF downloads
2,940





![[Table - 1]](#tbl_ijdvl_2006_72_5_334_27748_1.jpg){kind=link}
![[Figure - 1]](#fig_ijdvl_2006_72_5_334_27748_2.jpg){kind=link}
![[Table - 2]](#tbl_ijdvl_2006_72_5_334_27748_3.jpg){kind=link}
![[Table - 3]](#tbl_ijdvl_2006_72_5_334_27748_4.jpg){kind=link}
![[Table - 4]](#tbl_ijdvl_2006_72_5_334_27748_5.jpg){kind=link}
![[Table - 5]](#tbl_ijdvl_2006_72_5_334_27748_6.jpg){kind=link}
![[Table - 6]](#tbl_ijdvl_2006_72_5_334_27748_7.jpg){kind=link}