Translate this page into:
Viva questions from the IJDVL
2 Department of Dermatology, K J Somaiya Medical College and Research Centre, Sion, Mumbai, Maharashtra, India
Correspondence Address:
Vishalakshi Viswanath
Department of Dermatology, Rajiv Gandhi Medical College, Thane, Maharashtra
India
| How to cite this article: Viswanath V, Vasani R. Viva questions from the IJDVL. Indian J Dermatol Venereol Leprol 2016;82:114-119 |
Spotter[Figure - 1]
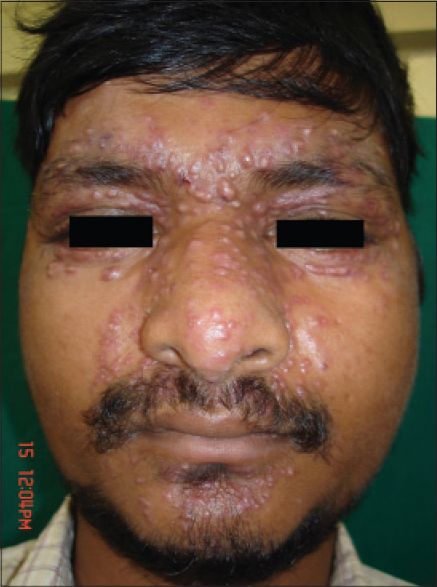 |
| Figure 1: Enumerate the differential diagnosis in this case. Courtesy: Rajiv Gandhi Medical College, Thane |
The differentials in this case include lupus miliaris disseminatus faciei, acne, granulomatous rosacea and sarcoidosis.
What are the synonyms for lupus miliaris disseminatus faciei?
The other terminologies are acne agminata, acnitis, Lewandowsky's rosaceiform eruption and facial idiopathic granulomas with regressive evolution.
How does lupus miliaris disseminatus faciei differ from its clinical mimickers?
- Lupus miliaris disseminatus faciei is characterized by the presence of reddish-brown symmetrical papulo-nodules which may show evidence of central necrosis. The lesions characteristically involve the eyelids, especially the lower eyelid
- In contrast to acne, there are no comedones and the lesions are not consistently related to the hair follicles. In contrast to lupus miliaris disseminatus faciei, acne has polymorphous lesions with comedones, inflammatory lesions, scars and pigmentary disturbances
- In lupus vulgaris, there is cutaneous hypersensitivity to tuberculin and isolation of mycobacterial DNA through polymerase chain reaction is possible, unlike lupus miliaris disseminatus faciei. Lupus vulgaris responds to antituberculous treatment while lupus miliaris disseminatus faciei does not
- Granulomatous rosacea has a background of erythema and telangiectasia and can have extra-facial lesions; these are uncommon in lupus miliaris disseminatus faciei
- Sarcoidosis can have associated systemic involvement while lupus miliaris disseminatus faciei does not
- Sudden eruption of dome-shaped papules and nodules in a leprosy patient with resistance to dapsone having characteristic epidermal Grenz zone and dermal spindle-shaped histiocytes characterizes histoid leprosy, whereas lupus miliaris disseminatus faciei shows perifollicular epithelioid cell granulomas with Langhans giant cells and occasionally caseation necrosis.
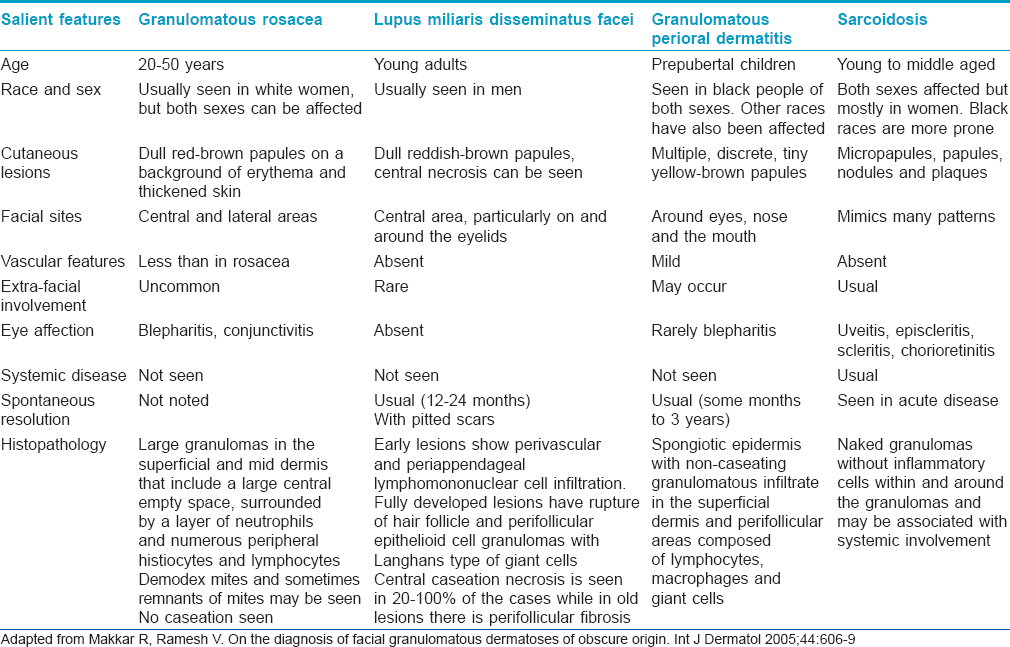
What are the idiopathic facial granulomatous eruptions and what are their differentiating features?
The salient differentiating features of idiopathic facial granulomatous eruptions are outlined in [Table - 1].
Therapeutic Paradox
What is a “therapeutic paradox?”
A “paradox” is defined as a “seemingly absurd or contradictory statement or proposition which when investigated may prove to be well founded or true.”
When a drug is given for the treatment of a particular condition, paradoxically, it is also responsible for the causation/exacerbation of the condition; this is called as a “therapeutic paradox.”
Enumerate some “therapeutic paradoxes” in dermatology
- Tumor necrosis factor alpha inhibitor-induced psoriasis
- Isotretinoin-induced acne fulminans
- Cetirizine-induced urticaria
- Chloroquine-induced lichenoid drug eruption
- Chloroquine-induced photosensitivity
- Topical calcineurin inhibitor-induced rosacea
- D-penicillamine-induced pseudoscleroderma
- Potassium iodide for erythema nodosum
- Clofazimine in erythema dyschromicum perstans
- Allergic contact dermatitis due to steroid moiety
- Antileprosy treatment as a trigger for reactions in leprosy
- Antiretroviral therapy and the immunological boost responsible for immune reconstitution inflammatory syndrome
- Intense pulsed light or laser-induced hypertrichosis
- Q-switched laser-induced darkening of tattoos.
Enumerate some paradoxes in clinical dermatology
- Allergic contact dermatitis due to toslyamide and formaldehyde induced resins in nail lacquer may occur on any part accessible to the nails, but dermatitis is not seen in the periungual area
- Oral exposure to nickel (through tap water/dental braces) may induce tolerance and reduced severity of the disease
- The “photo-hardening” effect of psoralen and ultraviolet A is utilized in treatment of photosensitive dermatoses
- Paraben and lanolin responsible for contact dermatitis produce false negative results on patch tests
- Vitamin B12 deficiency causes cutaneous hyperpigmentation but depigmentation of hair
- Development of leukoderma in melanoma may be seen in advanced stage of the disease with metastasis. Leukoderma indicates T-cell mediated immunological response against the target antigens present on both melanoma cells as well as normal melanocytes resulting in melanoma regression and vitiligo
- Itching is a consistent symptom in lichen planus, but paradoxically excoriations are conspicuously absent as the patient rubs rather than scratches to gain relief and hence the pathognomonic lesion morphology and Wickham's striae are preserved (Brocq's phenomenon)
- In cases of co-infection of leprosy and human immunodeficiency virus, the clinical, immunological and pathological features are same even in progressive human immunodeficiency virus disease and granuloma formation remains well preserved even with declining T-cell mediated immunity – the so-called “granuloma paradox.”
Adapted from and for further reading: Adya KA, Inamdar AC, Palit A. Paradoxes in dermatology. Indian Dermatol Online J 2013;4:133-42.
Trichoscopy Crossword
Identify the condition and solve the crossword puzzle [Figure - 2] based on the trichoscopic clues [Box 1]. Answers to the crossword puzzle are provided in [Figure - 3] on page 118.
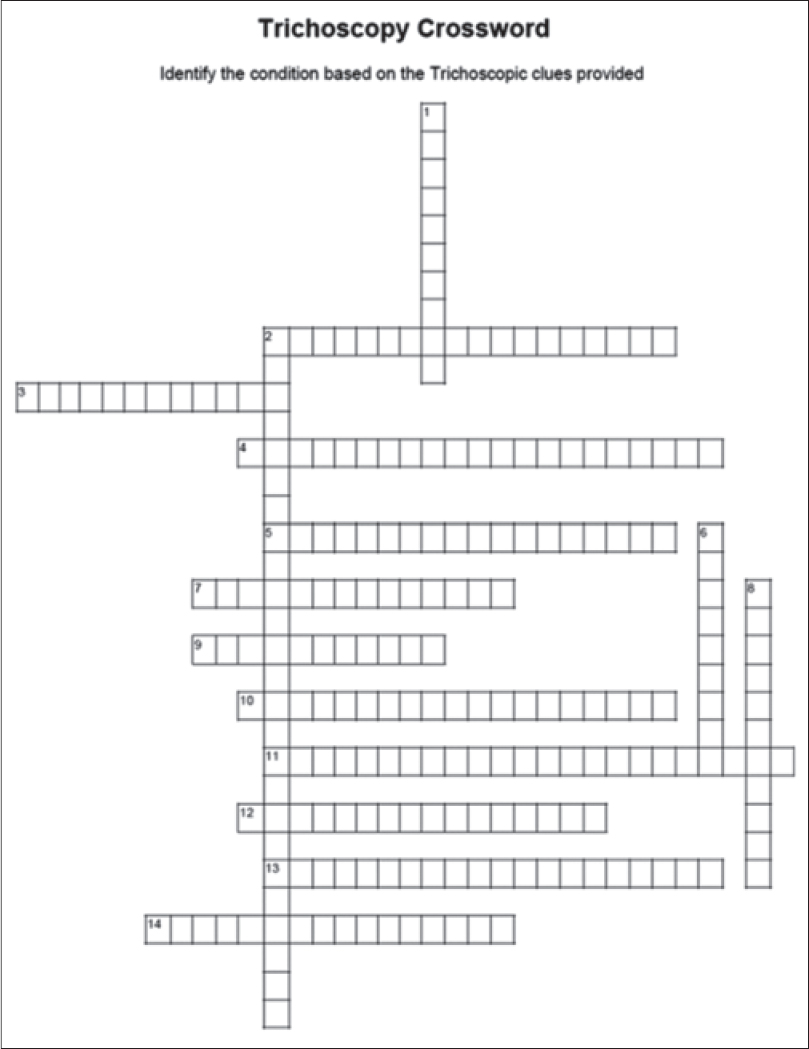 |
| Figure 2: Crossword puzzle (identify the condition based on the trichoscopic clues) |
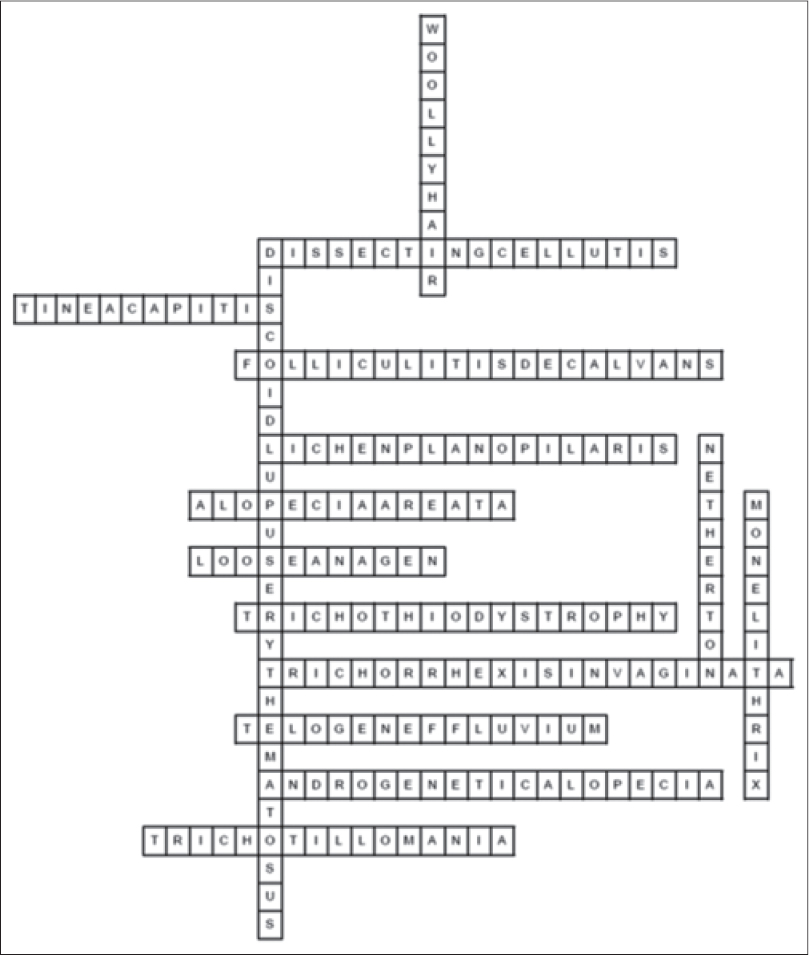 |
| Figure 3: Answer key to crossword puzzle |

Leser-Trelat Sign
What is sign of Leser-Trelat?
The sign of Leser-Trelat is characterized by the sudden eruption of numerous seborrheic keratoses, often with associated pruritus, or a rapid increase in their size within weeks or months. It is considered a marker of internal malignancy.
Enumerate the conditions wherein the sign of Leser-Trelat is found.
This sign has been described with various malignancies such as stomach cancer, gastrointestinal adenocarcinoma, lymphoma, leukemia, mycosis fungoides, Sezary syndrome, breast carcinoma, lung carcinoma, prostate carcinoma, laryngeal carcinoma and sarcomas (neurofibrosarcoma). The sign has been also reported in non-malignant situations as in heart transplant recipients, erythrodermic pityriasis rubra pilaris and human immunodeficiency virus infection; it can also occur in normal persons.
Kasabach–merritt Syndrome
What is Kasabach–Merritt syndrome or Kasabach–Merritt phenomenon?
Kasabach–Merritt phenomenon is characterized by a rapidly enlarging vascular tumor associated with thrombocytopenia, microangiopathic hemolytic anemia and a consumptive coagulopathy.
Which are the tumors associated with Kasabach–Merritt phenomenon?
Infantile hemangiomas, tufted angiomas, kaposiform hemangioendotheliomas, hemangiopericytomas and angiosarcomas can be associated with Kasabach–Merritt phenomenon. The location of these vascular tumors may be cutaneous, intrathoracic (mediastinal), abdominal (retroperitoneal or intrahepatic), pelvic or skeletal.
When should one suspect Kasabach–Merritt phenomenon?
Kasabach–Merritt phenomenon should be suspected in case of a rapidly enlarging vascular tumor associated with sudden appearance of ecchymoses which may extend beyond the margins of the tumor. There is a woody texture and the overlying inflammation may appear cellulitic. There is a bleeding diathesis that may manifest with hematuria, epistaxis and prolonged bleeding at sites of trauma. There is thrombocytopenia and activation of the fibrinolytic system. Due to the disseminated intravascular coagulation, there is consumption of clotting factors, low fibrinogen levels, elevated D-dimers, elevated prothrombin time and activated partial thromboplastin time.
What are the treatment options in Kasabach–Merritt phenomenon?
The treatment options depend on the severity of the coagulation defect, the site of tumor and the presence or absence of the physical effects of compression of surrounding tissues.
Initial conservative management with regular monitoring of platelets, clotting studies, fibrinogen and D-dimers should be done. Platelet transfusions and systemic corticosteroids should initially be used. Tumor reduction can be done with cytotoxic therapy, interferons. Surgical treatment (with pre-embolization) and radiotherapy can be considered in difficult cases.
Eosinophilic Fasciitis
What is the synonym of eosinophilic fasciitis?
This scleroderma-like syndrome is also termed as Shulman's syndrome. It is an uncommon connective tissue disease that may mimic and overlap with other sclerosing disorders such as morphea and lichen sclerosus.
Enumerate the trigger factors in eosinophilic fasciitis?
Eosinophilic fasciitis is triggered by various factors:
- Strenuous exercise and trauma are responsible in at least 66% of cases which are hypothesized to induce the antigenicity of the fascia and subcutis
- Arthropod bites, borreliosis and Mycoplasma arginini infection may be other triggers
- Drugs such as simvastatin, atorvastatin, ramipril and phenytoin may be responsible; ingestion of L-tryptophan may contribute to the eosinophilia -myalgia syndrome
- Neoplasia, autoimmune thyroiditis, eosinophilic colitis, hypercalcemia and amegakaryocytic thrombocytopenic purpura may be other factors associated with disease onset.
Describe the salient features of eosinophilic fasciitis
The characteristic clinical, histological and investigative findings include:
Clinical
Sudden-onset erythema, edema in the early phase and symmetrical woody induration of the distal extremities later are characteristic. There is a limitation of movement of the feet and hands. Occasionally, the face or abdomen can be affected and there may be superficial blistering and hemorrhage.
Histology
There is dermal sclerosis with inflammation and fibrosis extending to the subcutaneous tissue and deep fascia. The fascia is thickened and infiltrated with lymphocytes, plasma cells, histiocytes and eosinophils. Immunoglobulin G and C3 deposits can be demonstrated in the deep fascia. The inflammatory infiltrate consists of macrophages and predominantly CD8+ T-cells with eosinophils.
Investigations
Blood eosinophilia (up to 30%) is the predominant feature in most patients. Erythrocyte sedimentation rate may be elevated. There may be hyperglobulinemia, rarely, aplastic anemia and thrombocytopenia may be seen. Magnetic resonance imaging shows thickened deep fasciae on T1-weighted sequences and relatively increased signal intensity greater than that of muscle on fat-suppressed or fat-saturated T2-weighted sequences.
What are the overlapping and differentiating features of eosinophilic fasciitis and scleroderma?
The differentiation of eosinophilic fasciitis from deep morphea or overlapping cases with morphea or scleroderma may be problematic. Overlapping features of eosinophilic fasciitis and deep morphea are involvement of subcutaneous tissue, fascia, muscles, eosinophilia and positive antinuclear antibody and association with systemic sclerosis.
The features favoring the diagnosis of eosinophilic fasciitis are venous furrowing, prayer sign, the absence of sclerodactyly and Raynaud's phenomenon, prominent eosinophilia, hypergammaglobulinemia and the absence of nailfold capillary changes.
What are the treatment options?
Corticosteroids are the mainstay of treatment. Adjuvant modalities include hydroxychloroquine, methotrexate, cyclosporine and psoralen and ultraviolet A.
Photodynamic Therapy
What is the mechanism of photodynamic therapy?
Photodynamic therapy involves treatment with 5-aminolevulenic acid and various sources of light. There is absorption of 5-aminolevulenic acid by the rapidly proliferating epidermal and dermal cells and it is metabolized to the active photosensitizer, protoporphyrin IX, via the heme biosynthesis pathway. When protoporphyrin IX is activated by light, photodynamic reactions occur that lead to cell necrosis, apoptosis and remodulation.
What are the uses of topical photodynamic therapy in dermatology?
Topical photodynamic therapy with 5-aminolevulinic acid is a promising therapeutic modality for proliferative diseases such as actinic keratosis, Bowen's disease, in situ squamous cell carcinoma and superficial basal cell carcinoma. It has also been used in recalcitrant warts and warts in difficult to treat sites such as urethral condyloma acuminata. Photodynamic therapy is also being used in acne and as a photorejuvenation therapy.
Leopard Syndrome
What are the various clinical types of lentiginosis?
The term lentiginosis is used when there are numerous or extensive lentigenes, or they have a characteristic clinical pattern. The various clinical types are:
- Generalized lentiginosis
- Eruptive lentiginosis
- Zosteriform lentiginosis
- Centrofacial lentiginosis
- LEOPARD syndrome (lentigines, electrocardiographic abnormalities, ocular hypertelorism, pulmonary stenosis, abnormalities of genitalia, retardation of growth and deafness of sensorineural type)
- Peutz–Jeghers syndrome (periorificial lentigines, intestinal polyposis and soft-tissue malignancy)
- Cronkhite–Canada syndrome (generalized gastrointestinal polyposis, cutaneous hyperpigmentation, hair loss and nail atrophy)
- <>
- Carney complex or LAMB syndrome (lentigines, atrial myxomas, mucocutaneous myxomas and blue nevi).
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Fulltext Views
9,324
PDF downloads
2,748





![[Figure - 1]](#fig_ijdvl_2016_82_1_114_172911_f1.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2016_82_1_114_172911_t4.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2016_82_1_114_172911_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2016_82_1_114_172911_f3.jpg){kind=link}