Translate this page into:
Viva questions from the IJDVL
2 Department of Dermatology, K J Somaiya Medical College and Research Centre, ion, Mumbai, Maharashtra, India
Correspondence Address:
Vishalakshi Viswanath
Department of Dermatology, Rajiv Gandhi Medical College, Thane, Maharashtra
India
| How to cite this article: Viswanath V, Vasani R. Viva questions from the IJDVL. Indian J Dermatol Venereol Leprol 2015;81:97-101 |
Spotter
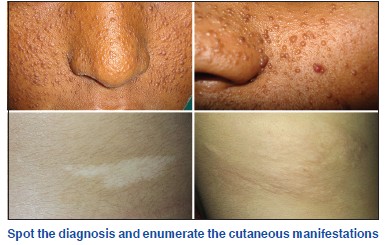
What are the other names for tuberous sclerosis?
Bourneville′s disease; Pringle disease; EPILOIA (epilepsy, low intelligence, adenoma sebaceum).
What is the inheritance of tuberous sclerosis?
Inheritance is autosomal dominant with variable expression and approximately 60-70% of tuberous sclerosis complex (TSC) is attributable to a new mutation.
There are two genes: TSC1 on chromosome 9q34, which encodes for the protein hamartin and TSC2 on chromosome 16, which encodes for the protein tuberin. These are tumor suppressor genes which when deficient can result in mammalian target of rapamycin (mTOR) disinhibition and abnormal proliferation of tissues, resulting in hamartomas.
What are the cutaneous features of tuberous sclerosis?
A. Ash leaf macule
- Seen in 95% of the cases
- Present at birth or appears in the neonatal period. Presence of three or more hypomelanotic macules at birth should raise the suspicion of tuberous sclerosis complex.
- One to twenty, 0.5-3 cm diameter off-white macules. Some are oval at one end and taper to a point at the other and are hence called lanceolate or ash leaf macules. Confetti or polygonal macules with smooth or irregular margins can also occur.
- Histopathology shows normal number of melanocytes but absent melanosomes.
- They fade or disappear in adulthood. New lesions can appear as age increases.
B. Facial angiofibromas
- Seen in 88-100% of cases.
- Present at 2-5 years. Occasionally can arise in adulthood.
- The lesions are heralded by excessive facial flushing in infancy. Later 1-3 mm, pink to red papules that have a smooth surface occur on the central face, and often concentrate around alar grooves, extending symmetrically over the nose, cheeks, nasal opening and chin with relative sparing of the upper lip and lateral face.
- Plump, spindle-shaped or stellate fibroblasts are seen in the dermis among increased numbers of dilated blood vessels. Collagen fibers are oriented in onion skin pattern around follicles and blood vessels.
- During puberty, they may grow in size and number. In adulthood, they tend to be stable in size but redness may gradually diminish.
C. Fibrous facial plaque
- Seen in 2.5-55.5% cases.
- Congenital or appears shortly after birth.
- Irregular, soft to firm, of normal skin color, red or hyperpigmented in dark colored individuals. They are found on scalp, cheeks and elsewhere on the face.
- These are connective tissue nevi of collagen type without vascular dilatation.
- These do not spontaneously regress.
D. Shagreen patch
- Seen in 12.3-83% cases.
- May present in infancy but can present later.
- Skin colored leathery plaque with prominent follicular openings resembling an orange peel. Commonly seen over the lumbo-sacral area.
- Sclerotic bundles of collagen are found in the reticular dermis. Elastic fibers are typically reduced or absent.
- Do not spontaneously regress.
E. Ungual fibromas/Koenen′s tumors
- Found in 6.2-66.6% of adults.
- Usually appear after the first decade. May sometimes appear in middle age.
- They are 1 mm to 1 cm in diameter and arise from under the proximal nail fold or under the nail plate. They arise from the nail matrix and cause a longitudinal groove and sometimes a groove forms without a papule. Subungal fibromas can be seen through the nail plate as red or white oval lesions or red papules emerging from the distal nail plate causing distal subungal onycholysis.
- Histology shows angiofibromas with prominent hyperkeratosis and a variable increase in vascularity.
- Do not spontaneously regress.
F. Other dermatological features include
- Fibromas around the teeth and even on the tongue.
- Dental pits particularly affecting adult teeth.
- Skin tags called molluscum fibrosum pendulum especially around the neck.
- Café au lait macules.
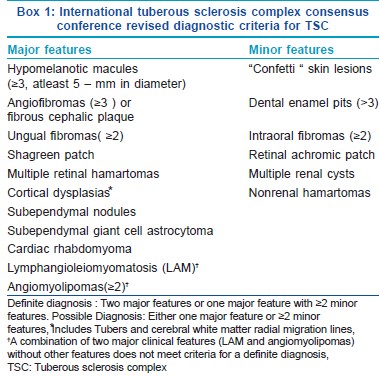
What are the criteria for diagnosis of tuberous sclerosis?
International TSC Consensus Conference Revised Diagnostic criteria for tuberous sclerosis complex laid out in 2012 are as in Box 1.
Enumerate various types of adenoma sebaceum
- Balzer type - There are only slight changes in the sebaceous glands.
- Pringle type - There is enormous hyperplasia of the sebaceous glands.
- Hallopeau-Leredde type - There is a predominance of the fibrous tissue with a verrucous surface.
- Mixed type - Mixed appearance where one lesion shows great hyperplasia of the sebaceous gland and neighboring lesion shows no sebaceous change.
- Epithelioma adenoides cysticum (misnomer - as they are actually trichoepitheliomas).
How has the name tuberous sclerosis been derived?
The name is derived from Latin tuber and the Greek skleros (hard). Tubers are potato-like nodules of glial proliferation and are characteristic central nervous system lesions of tuberous sclerosis complex. They may occur anywhere in the cerebral cortex, basal ganglia and ventricular walls (subependymal nodules) and their number and size correlate with the clinical features of seizures and mental retardation. Cortical tubers are often isodense with normal brain tissue and are best detected by magnetic resonance imaging. Subependymal nodules may calcify and are readily detectable with computerized tomography scanning. Fifty percent of plain skull X-rays taken in later childhood reveal bilateral areas of calcification in the brain.
What is the role of mammalian target of rapamycin inhibitors in tuberous sclerosis complex?
Tuberous sclerosis is an autosomal dominant tumor syndrome that results from mutations in the tumor suppressor hamartin (TSC1) or tuberin (TSC2). Hamartin and tuberin normally suppress mTOR, which increases cell cycle progression when it is released from negative regulation. The loss of tumor suppressive function in tuberous sclerosis complex leads to the formation of multiple tumors of the internal organs and skin.
Rapamycin or sirolimus belongs to a novel group of molecules known as mTOR inhibitors and is approved for use in renal transplantation and drug-eluting stents in United States. Two derivates of rapamycin, everolimus and temsirolimus are in clinical development for similar indications. Rapamycin forms an inhibitory complex with the immunophilin FKBP12 and corrects the aberrant signaling in a variety of pathways that regulate cell growth and apoptosis. It also inhibits vascular endothelial growth factor which is a proangiogenic molecule. Oral rapamycin can improve the renal, brain and skin lesions in tuberous sclerosis complex. Topical rapamycin has been shown to be effective in the treatment of facial angiofibromas without significant adverse effects.
Acrodermatitis Enteropathica
What is the etiopathogenesis of acrodermatitis enteropathica?
Acrodermatitis enteropathica is a rare, autosomal recessive disorder classically characterized by zinc deficiency due to defective absorption. It is associated with ocular, gastrointestinal, and mucocutaneous symptoms with variable frequency, and can be fatal without zinc therapy.
What are the clinical features of acrodermatitis enteropathica?
Infants with acrodermatitis enteropathica present with the classic triad of diarrhea, skin rash with alopecia and extreme irritability with depression. It is common in premature babies due to insufficient body stores of zinc as well as high zinc requirements at the time of weaning. It is rare in breastfeeding infants because of zinc binding ligand in breast milk which generally protects breastfed infants against this condition by increasing zinc bioavailability. Onset is approximately 1-2 weeks after weaning or 4-10 weeks of age in bottle-fed infants. Infants with acrodermatitis enteropathica have a classical erythematous, scaly, psoriasiform and sometimes vesiculo-erosive eruption located periorificially (i.e. around the mouth, eyes and genital area). Refractory diaper dermatitis may be the presenting complaint. Nail changes consisting of paronychia as well as dystrophy of the nail plate may occur. Superadded staphylococcal and candidal infections are very common, and candida may not respond to topical therapy until zinc deficiency is corrected.
How is acrodermatitis enteropathica diagnosed?
- Samples for determining zinc levels must be carefully collected in special acid washed or plastic tubes so that exogenous zinc present on ordinary glassware will not contaminate the specimen. Serum zinc levels are usually less than 50 μg/dl.
- Low alkaline phosphatase levels can be a valuable marker of zinc deficiency even when the plasma zinc levels are normal as it is a zinc dependent enzyme.
What is the treatment for acrodermatitis enteropathica?
Treatment is with oral zinc, 3-5 mg/kg/day. Life-long zinc supplementation is required with a diet rich in zinc in the form of unmilled cereals, beans, cheese, whole wheat bread and animal proteins like lean red meat is necessary to prevent recurrences.
Parthenium Dermatitis
What are the clinical patterns of parthenium dermatitis?
Clinical types:
- Classic airborne contact dermatitis
- Chronic actinic dermatitis
- Mixed pattern (airborne-photosensitive pattern)
- Exfoliative dermatitis
- Prurigo nodularis-like eruption
- Photosensitive lichenoid eruption
- Actinic prurigo-like lesions.
Secondary Comedones After Waxing
What are the conditions in dermatology where we see comedones?
- Acne
- Porokeratotic eccrine osteal and dermal duct nevus
- Familial dyskeratotic comedones
- Darier′s disease
- Acne mechanica
- Comedone nevus
- Solar comedones
- Follicular mycosis fungoides
- Systemic amyloidosis.
Xeroderma Pigmentosum
What are the salient features of genetic disorders commonly associated with photosensitivity?
The genetic disorders commonly associated with photosensitivity include:
- Cockayne syndrome
- Xeroderma pigmentosum (XP)
- Rothmund-Thomson syndrome
- Bloom syndrome
- Ataxia telangiectasia.
The salient clinical features are outlined in [Table - 1].
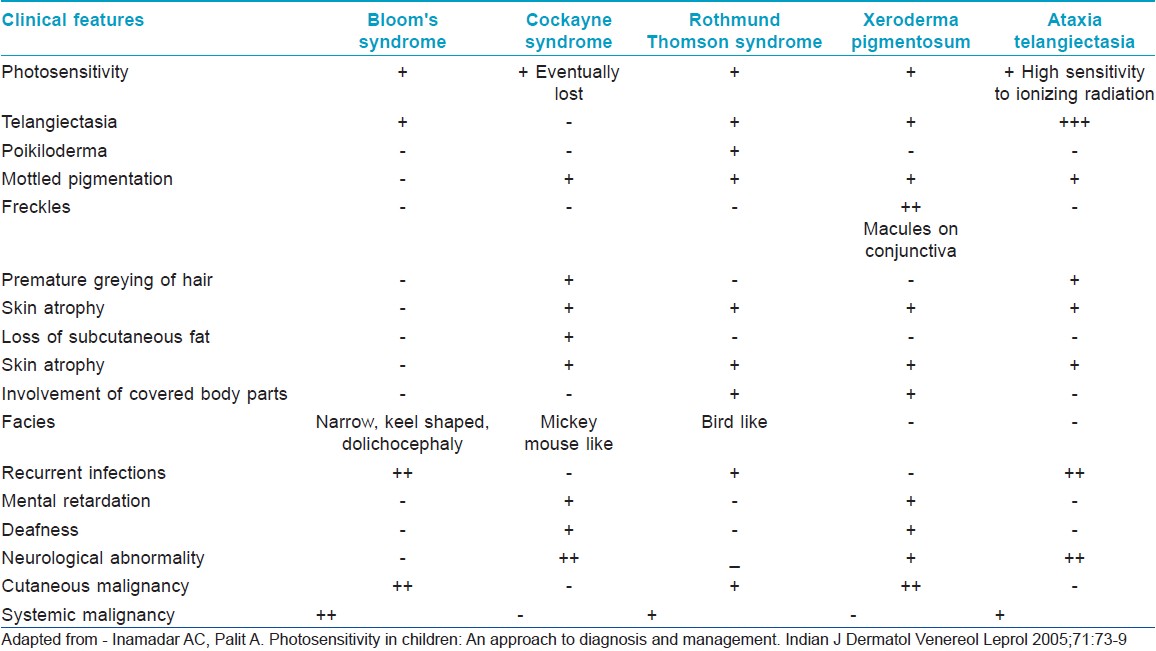
What is the inheritance pattern in xeroderma pigmentosum?
Xeroderma pigmentosum is an autosomal recessive genetic disorder of DNA repair characterized by cutaneous and ocular photosensitivity. Mutations arise most commonly in XPA (25%), XPC (25%), POLH1 (21%) or XPD (15%) genes.
Which tumors are associated with xeroderma pigmentosum?
Basal cell carcinoma, squamous cell carcinoma, melanoma, keratoacanthoma, angiosarcoma and fibrosarcoma can occur. Tumors tend to be multiple and are frequently associated with multiple metastases.
What are the treatment options in xeroderma pigmentosum?
Strict photoprotection (clothing, hats, sunscreens) and care of eyes (artificial tears, sunglasses) is mandatory. Early and adequate excision of all tumors is essential. Topical 5-fluorouracil may be useful for early or premalignant lesions. Chemical peeling and dermabrasion can be done. Prophylactic therapy with systemic retinoids may reduce the onset of skin cancers. The microbial enzyme T4 endonuclease V regularly applied as a topical liposome lotion has also been reported to be useful.
Pruritus in Hiv
Enumerate various causes of pruritic papular eruptions in HIV
Insect bite reaction or papular urticaria, scabies, pityrosporum folliculitis, demodex folliculitis, bacterial and viral folliculitis, eosinophilic pustular folliculitis and drug reactions present with severely pruritic papules.
Blaschko Lines
What are Blaschko′s lines?
The lines of Blaschko represent a form of "mosaicism." They represent the routes of ectodermal cell migration from the neural crest.
These lines are in the form of transverse bands on the trunk, S-shaped on the lateral trunk and V-shaped in the middle of the back. The lesions follow lines parallel with the axis of the limb on the arms and legs. They are spiral shaped on the scalp, vertical in the mid-face, and extend laterally from the angles of the mouth. The lines do not cross the anterior truncal midline but run along it. Most epidermal nevi follow Blaschko′s lines.
Name some cutaneous disorders occurring along Blaschko′s lines
Various inherited and acquired disorders of hyperpigmentation have been described along Blaschko′s lines. Many nevoid and acquired skin conditions may follow these lines including incontinentia pigmenti, focal dermal hypoplasia, epidermal nevus, sebaceous nevus, lichen nitidus, lichen planus, lichen striatus, lupus erythematosus, vitiligo, and psoriasis. Darier′s disease, Hailey-Hailey disease, porokeratosis, atrophoderma of Moulin, and "adult Blaschkitis" or BLAISE (Blaschko linear acquired inflammatory skin eruption) can also occur in this pattern. Other uncommon entities that may show this pattern include lupus erythematosus, fixed drug eruption, chronic graft-versus-host disease, mycosis fungoides and blaschkoid mastocytosis.
INTERFERONS
Enumerate the uses of interferons in dermatology
Tumors: Adjuvant treatment of Stage II and III melanoma, cutaneous T- and B-cell lymphoma, AIDS-related Kaposi sarcoma, basal cell carcinoma, squamous cell carcinoma, actinic keratosis, plexiform neurofibroma.
Non-tumor indications: Genital warts, herpes infection, leishmaniasis, mastocytosis, Behcet′s disease, hemangiomas, Gorham-Stout syndrome, atopic dermatitis, keloids.
Fabry′s Disease
Classify angiokeratomas
Angiokeratomas are classified into either localized or diffuse forms. The localized form includes: (1) solitary papular type, which can manifest on any body part as a single lesion; (2) Fordyce type, which occurs on the scrotum or the vulva; (3) Mibelli type, which presents as hyperkeratotic vascular lesions over the bony prominences of the hands and feet; and (4) angiokeratoma circumscriptum neviforme, which is a congenital unilateral zosteriform verrucous lesion of the lower extremities.
The systemic or diffuse form, angiokeratoma corporis diffusum, can involve the entire body and is associated with lysosomal storage disease including Fabry disease, gangliosidosis, aspartylglucosaminuria, fucosidosis, β-mannosidosis, sialidosis, galactosialidosis, and Kanzaki disease.
Fox-Fordyce Disease
What is the other name of Fox-Fordyce disease? Describe the clinical features.
Fox-Fordyce disease is also known as apocrine miliaria. It is a rare condition seen in adolescent women, over the apocrine gland-bearing areas, like the axillae, groin, areolae and inframammary creases in decreasing order of frequency. There may be intense itching that may be aggravated by emotional stimuli. Itching is common in the axillae. Clinical manifestations are in the form of skin-coloured, or slightly pigmented, dome-shaped, follicular papules. It may be followed by hair loss in the axillae. The disease has a prolonged course; it may remit in pregnancy or persist until menopause.
What are the treatment options for Fox-Fordyces disease?
Topical steroids, adapalene, benzoyl peroxide, pimecrolimus, clindamycin, oral retinoids, oral contraceptives can be tried. Laser therapy and surgical interventions can be tried in resistant cases.
Fulltext Views
6,985
PDF downloads
3,076





![[Table - 1]](#tbl_ijdvl_2015_81_1_97_148618_t3.jpg){kind=link}