Translate this page into:
Whole exome sequencing in a multi-generation family from India reveals a genetic variation c.10C>T (p.Gln4Ter) in keratin 5 gene associated with Dowling–Degos disease
2 Genomics and Molecular Medicine, CSIR Institute of Genomics and Integrative Biology, New Delhi, India
3 Department of Dermatology, Seth GSMC and KEM Hospital, Mumbai, Maharashtra, India
Correspondence Address:
Vinod Scaria
CSIR Institute of Genomics and Integrative Biology, Mathura Road, New Delhi - 110 025
India
| How to cite this article: Virmani N, Vellarikkal SK, Verma A, Jayarajan R, Sakhiya J, Desai C, Sivasubbu S, Scaria V. Whole exome sequencing in a multi-generation family from India reveals a genetic variation c.10C>T (p.Gln4Ter) in keratin 5 gene associated with Dowling–Degos disease. Indian J Dermatol Venereol Leprol 2018;84:344-346 |
Sir,
Dowling–Degos disease (OMIM: 179850) is a rare genodermatosis characterized by reticulate pigmentation of the flexures.[1] This disease is inherited in an autosomal dominant pattern,[2] with onset typically in early adulthood. The clinical presentation is with development of hyperpigmented, reticular macules on the face, trunk, axillae and groin.[1],[3] The mutations causing the disease are usually found in the keratin 5 (KRT5)[4] gene and rarely in the protein O-fucosyl transferase 1 (POFUT1)[5] and protein O-glucosyltransferase 1 (POGLUT1) genes.[6] POGLUT1 and POFUT1 are components of the Notch signaling pathway, involved in the differentiation and migration of cells. Mutations in these genes affect the skin more than the hair.[5] Only a few cases suffering from this disease have been described previously from India, as it is a rare disorder with an unknown population frequency.[7],[8] In the current report, we emphasize the utility of whole exome sequencing in the molecular characterization of the genetic defect, in a rare familial case of a reticular pigmentation disorder.
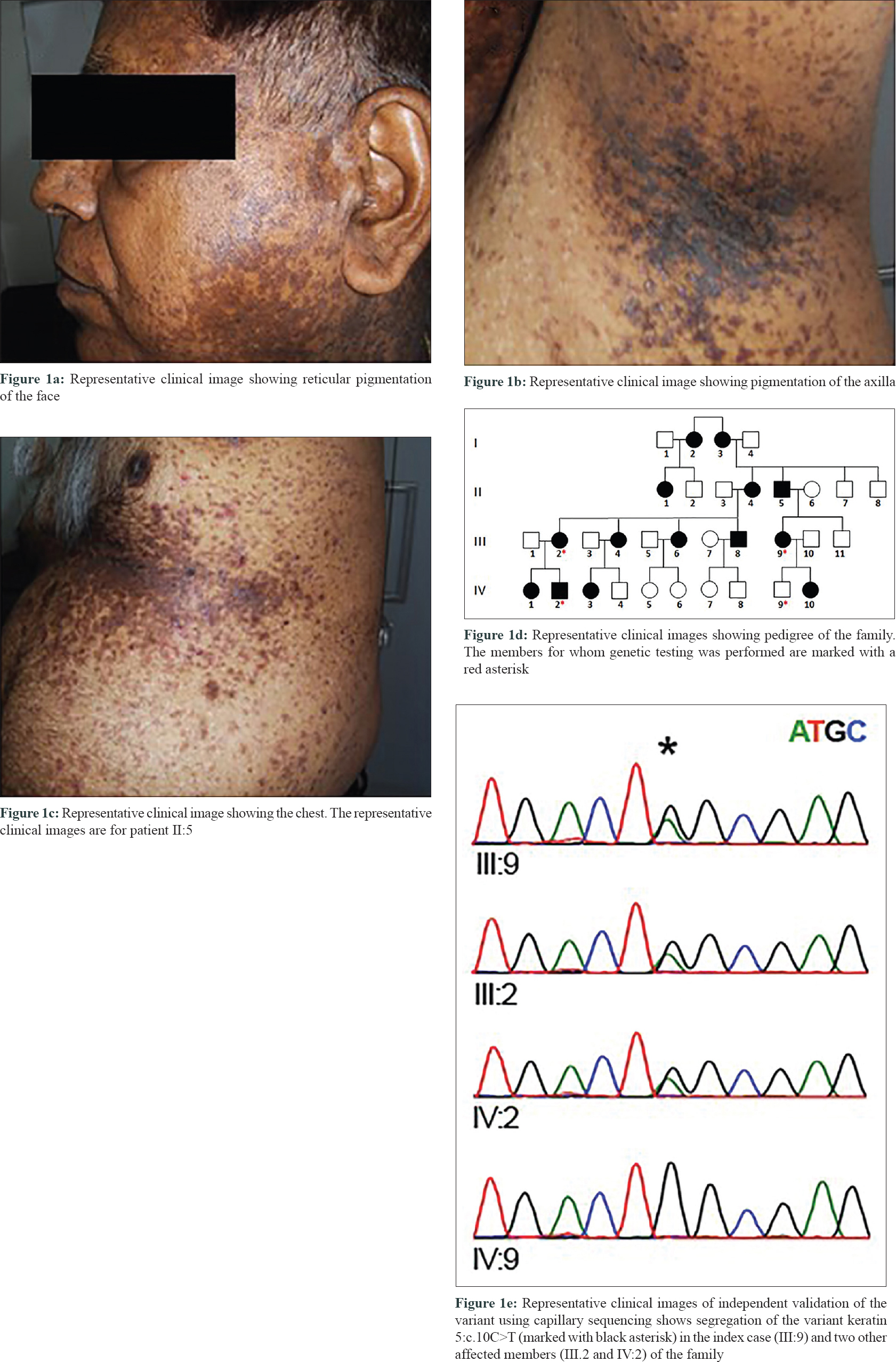
The multi-generational family comprised over 30 individuals in four generations, with 14 members suffering from a reticular pigmentation abnormality [Figure - 1]a,[Figure - 1]b,[Figure - 1]c,[Figure - 1]d affecting the face, axillae and the trunk. Some members of the family have been previously reported in a recent case series.[7] The Proband (III.9) was a 42-year-old female who presented with multiple hyperpigmented papules and macules localized to the face, neck and abdomen. Acantholysis was seen on histopathology. Her father (II.5) had multiple wide-spread itchy erythematous papules and plaques involving the face, upper and lower limbs and trunk [Figure - 1]a,[Figure - 1]b,[Figure - 1]c,[Figure - 1]d. Her daughter (IV.10) had similar lesions, while her son (IV.9) was asymptomatic. Another member (III.2) of the family had multiple erythematous and hyperpigmented itchy papules and patches over the face, abdomen and limbs, clinically suspected to be a case of Galli–Galli disease. Her mother (II.4) and both the affected children (IV.1 and IV.2) showed similar features.
 |
| Figure 1 |
Because there were three genes typically mutated in Dowling–Degos disease and KRT5 mutation screening was not readily available in India, we attempted whole exome sequencing to characterize the genetic variation. In addition, our selection of the technique was guided by the fact that reticular pigmentation disorders have overlapping phenotypes and genes involved. The study was approved by the Institutional human ethics committee of CSIR-IGIB (IHEC proposal no. 8). DNA was isolated from whole blood using salt-precipitation method and whole exome sequencing (WES) was performed for one affected individual following manufacturer's instructions (Illumina Inc., USA). WES analysis revealed a G to A transition (chr12:g.52914071G>A) in exon 1 of KRT5 gene, which also corresponds to C to T change at position 10 in KRT5 cDNA (c.10C>T). The mutation resulted in the substitution of glutamine by a stop codon at fourth residue (p. Gln4Ter). This variant was not present in 1000 genome, ExAC and al mena [9] datasets and was also absent in the internal control database of South Asian Genomes and Exomes. The variant was present in ClinVar (RCV000056543.1) with no clinical assertions provided. We also attempted to systematically curate literature evidence for genetic variants associated with Dowling–Degos disease. Detailed literature analysis revealed that the genetic variant KRT5:c.10C>T was previously reported from India [8] and China [10] in patients suffering from Dowling–Degos disease and presenting with similar clinical features. The variant was further confirmed by capillary sequencing in the index case (III:9) and two other affected members (III.2 and IV: 2), and was found to be absent in one unaffected member (IV:9) [Figure - 1]e.
The mode of inheritance, familial segregation, functional consequences and the extreme rarity of the variation in population frequency databases provides ample evidence that the variation c.10C>T [8],[10] in KRT5 is associated with Dowling–Degos disease. Apart from confirming the association of the variant through familial segregation, our analysis suggests that the variant could potentially be quite prevalent in the population, given another report from an unrelated family.[4] Therefore, we suggest that a systematic screening for the mutation could potentially benefit a large number of patients and families. Given the cost-effectiveness, speed and ease of use, we propose that whole-exome sequencing could be of clinical utility for effective molecular diagnosis in rare cases of genodermatoses, especially in cases where overlapping clinical features could preclude an accurate diagnosis.
Acknowledgements
Authors acknowledge funding for the study from the Council of Scientific and Industrial Research, India and thank Genomics for Understanding Rare Diseases India Alliance Network (GUaRDIAN) for the support.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given his consent for her images and other clinical information to be reported in the journal. The patient understands that name and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Funding from the Council of Scientific and Industrial Research (CSIR), India through Grant No. BSC0212 (Wellness Genomics Project). The funding agency did not have any role in study design; data collection, analysis, and interpretation; writing the report; and the decision to submit the report for publication.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Sardana K, Goel K, Chugh S. Reticulate pigmentary disorders. Indian J Dermatol Venereol Leprol 2013;79:17-29.
[Google Scholar]
|
| 2. |
Crovato F, Nazzari G, Rebora A. Dowling-Degos disease (reticulate pigmented anomaly of the flexures) is an autosomal dominant condition. Br J Dermatol 1983;108:473-6.
[Google Scholar]
|
| 3. |
Mohan L, Bhatiya PS, Mishra R, Singh KK. Dowling-Degos disease. Indian J Dermatol Venereol Leprol 1991;57:251.
[Google Scholar]
|
| 4. |
Betz RC, Planko L, Eigelshoven S, Hanneken S, Pasternack SM, Bussow H, et al. Loss-of-function mutations in the keratin 5 gene lead to Dowling-Degos disease. Am J Hum Genet 2006;78:510-9.
[Google Scholar]
|
| 5. |
Li M, Cheng R, Liang J, Yan H, Zhang H, Yang L, et al. Mutations in POFUT1, encoding protein O-fucosyltransferase 1, cause generalized Dowling-Degos disease. Am J Hum Genet 2013;92:895-903.
[Google Scholar]
|
| 6. |
Basmanav FB, Oprisoreanu AM, Pasternack SM, Thiele H, Fritz G, Wenzel J, et al. Mutations in POGLUT1, encoding protein O-glucosyltransferase 1, cause autosomal-dominant Dowling-Degos disease. Am J Hum Genet 2014;94:135-43.
[Google Scholar]
|
| 7. |
Desai CA, Virmani N, Sakhiya J, Khopkar U. An uncommon presentation of Galli-Galli disease. Indian J Dermatol Venereol Leprol 2016;82:720-3.
[Google Scholar]
|
| 8. |
Verma S, Pasternack SM, Rütten A, Ruzicka T, Betz RC, Hanneken S, et al. The first report of KRT5 mutation underlying acantholytic Dowling-Degos disease with mottled hypopigmentation in an Indian family. Indian J Dermatol 2014;59:476-80.
[Google Scholar]
|
| 9. |
Koshy R, Ranawat A, Scaria V. Al mena: A comprehensive resource of human genetic variants integrating genomes and exomes from Arab, Middle Eastern and North African populations. J Hum Genet 2017;62:889-94.
[Google Scholar]
|
| 10. |
Guo L, Luo X, Zhao A, Huang H, Wei Z, Chen L, et al. Anovel heterozygous nonsense mutation of keratin 5 in a Chinese family with Dowling-Degos disease. J Eur Acad Dermatol Venereol 2012;26:908-10.
[Google Scholar]
|
Fulltext Views
1,539
PDF downloads
1,560





![[Figure - 1]](#fig_ijdvl_2018_84_3_344_228984_f1.jpg){kind=link}