Translate this page into:
Ichthyosis bullosa of Siemens: Response to topical tazarotene
Correspondence Address:
S V Rakhesh
Department of Dermatology and STD, Academy of Medical Sciences, Pariyaram, Kannur 670503, Kerala
India
| How to cite this article: Rajiv S, Rakhesh S V. Ichthyosis bullosa of Siemens: Response to topical tazarotene. Indian J Dermatol Venereol Leprol 2006;72:43-46 |
Abstract
In 1937, Siemens described a Dutch family with superficial blistering, flexural hyperkeratosis, and characteristic mauserung appearance. Since then, less than 20 kindreds with this condition have been described in the English dermatologic literature. A 14-year-old boy presented with history of recurrent blistering and peeling of skin since the age of 1 month, predominantly seen over limbs and trunk, often associated with secondary infection. His mother also had similar symptoms from childhood. On examination, the child had typical mauserung peeling of the skin and dirty gray hyperkeratosis in a rippled pattern over flexures. Skin biopsy from the boy showed intracorneal blistering with epidermolytic hyperkeratosis in the upper spinous layers. The typical history and clinical features along with characteristic histological findings confirmed our diagnosis of ichthyosis bullosa of Siemens. It must be differentiated from other conditions with epidermolytic hyperkeratosis and skin peeling, such as bullous ichthyosiform erythroderma of Brocq and peeling skin syndrome. Our patient responded well to 0.05% topical tazarotene gel over four weeks.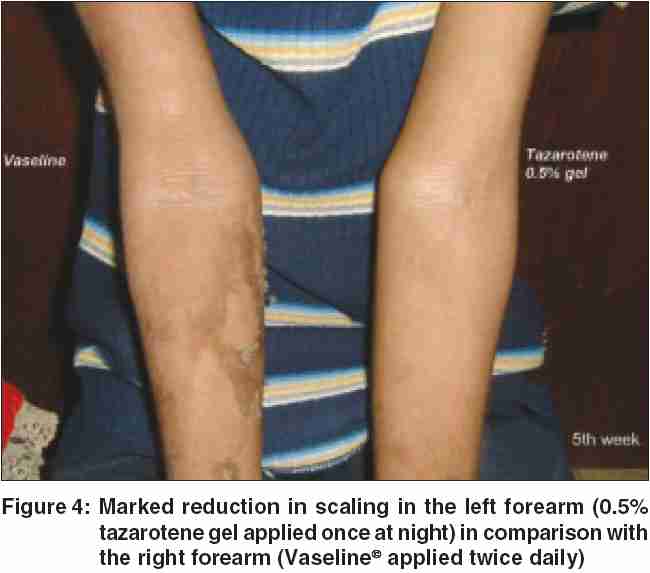 |
|
 |
|
 |
|
 |
|
INTRODUCTION
In 1937, Hermann Werner Siemens, a pioneer in the field of genetic medicine, described a Dutch family suffering from a hitherto unknown skin disorder characterized by superficial blistering, flexural hyperkeratosis, and a characteristic mauserung or "molting" appearance.[1] He called it "ichthyosis bullosa" and delineated it from bullous congenital ichthyosiform erythroderma (BCIE) described earlier by Brocq in 1907.[1] Since then, this entity had fallen into oblivion until 49 years later, when in 1986, Traupe et al. described the clinical, histological, and ultrastructural details of this disease in a Northwestern German family.[2] Recent molecular studies have demonstrated a keratin 2e (K2e) mutation in families with the disease,[3] further distinguishing it from BCIE,[4] where mutations have been observed in the keratin-1 and -10 genes. We report yet another family affected with this rare entity.
CASE REPORT
0Case 1
A 14-year-old boy born of a non-consanguineous marriage was seen in our outpatient department with a history of recurrent blistering and peeling of the skin since the age of 1 month. He was apparently normal at birth. The blistering occurred predominantly over the trunk and lower limbs and often became secondarily infected, causing considerable discomfort. Although the problem of blistering had progressively improved with age, the problem of skin peeling increased over the years. The boy was otherwise healthy. He had never suffered from erythroderma and there was no history of hyperhidrosis of the palms and soles. His mother suffered from similar complaints from her childhood, but his other sibling was normal.
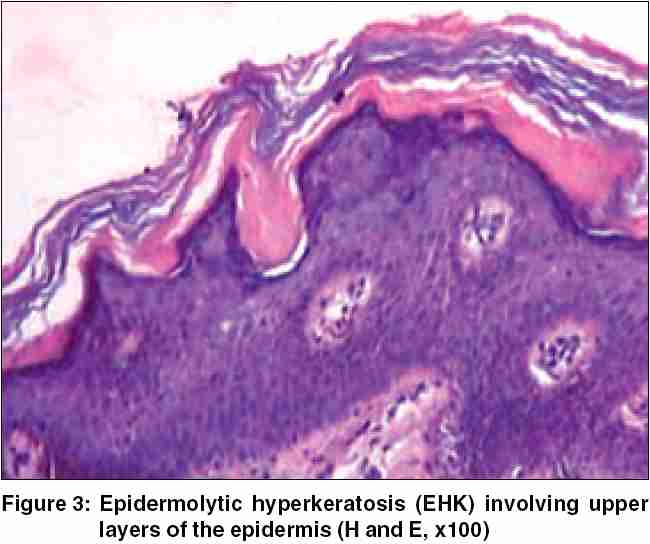
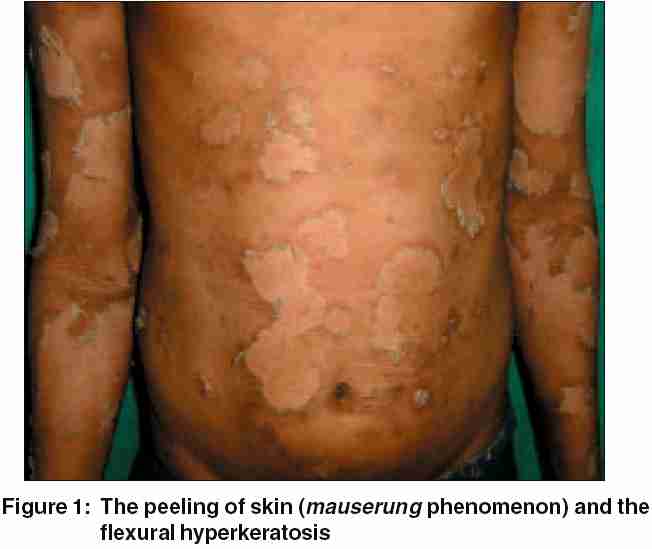
On physical examination, the boy displayed multiple, variously sized, circumscribed areas of peeling of the skin [Figure - 1], similar to the mauserung phenomenon described by Siemens.[1] This phenomenon was observed over most of his body, with relative sparing of the face, anterior chest, and dorsal aspect of fingers. The denuded areas of skin looked apparently normal, with no erythema or hyperkeratosis. The flexural aspects of the extremities and the anterior abdomen showed dirty gray hyperkeratosis with a typical rippled pattern. A fresh, flaccid blister set on a non-erythematous background was noted on the inner aspect of the thigh [Figure - 2]. His palms, soles, hair, nails, and mucosae were normal. The differential diagnoses considered at this juncture were ichthyosis bullosa of Siemens (IBS), familial peeling skin syndrome (PSS), and bullous congenital ichthyosiform erythroderma (BCIE). An overlap between IBS and PSS was also kept in mind.
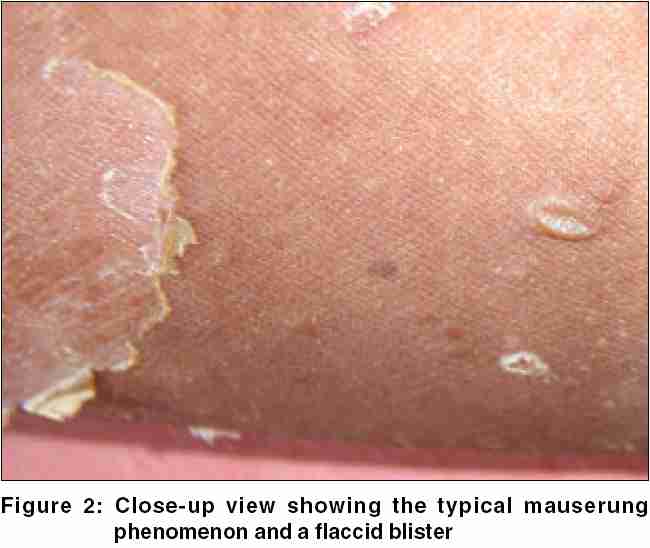
Histological examination of the specimen taken from the margin of an area exhibiting the mauserung phenomenon disclosed intracorneal blistering with hyperkeratosis [Figure - 3]. The prominent granular layer and the upper spinous cell layers showed coarse clumping of keratohyaline granules. The upper spinous cells also showed a few vacuolated keratinocytes. Apart from the acanthotic changes, the rest of the epidermis as well as the underlying dermis were unremarkable. The histological changes were thus compatible with the phenomenon of epidermolytic hyperkeratosis (EHK), principally involving the upper layers of the epidermis.
Our patient responded remarkably well to topical treatment with 0.05% tazarotene gel, with reduction in blistering and scaling noted at the end of 4 weeks. He was asked to apply white, soft paraffin twice daily on his right arm, whereas he was given 0.05% tazarotene gel to be applied at nighttime on his left arm. The flexural hyperkeratosis and the mauserung appearance were noted to be significantly less in the arm tazarotene was applied at the end of 4 weeks [Figure - 4]. He is on regular follow-up.
Case 2
The patient′s 35-year-old mother gave a similar history of blistering and peeling of the skin since 1 month of age. As in her son, the blistering decreased with age and eventually ceased. She noticed thickening of her skin predominantly over the sites of friction. On examination, her upper and lower extremities showed rippled flexural hyperkeratosis along with the typical mauserung type of peeling. However, the patient refused a skin biopsy.
DISCUSSION
In 1902, Brocq described bullous ichthyotic erythroderma and distinguished blistering from nonblistering congenital ichthyosiform erythroderma.[5] The characteristic histological picture of hyperkeratosis with lysis and tonofilament clumping in the granule cell layer has led it to be called EHK. Today, the diagnosis of EHK remains imprecise, with many variations being described.[4] Traupe et al. discussed three types of EHKs: bullous congenital ichthyosiform erythroderma of Brocq, ichthyosis bullosa of Siemens, and ichthyosis hystrix of Curth-Macklin.[6] DiGiovanna and Bale (1994) separated the various clinical presentations of EHK into two primary types, the "NPS types" (those without severe palm/sole hyperkeratosis) and the "PS types" (those with severe palm/sole hyperkeratosis).[7] The two primary types were subdivided further into three subtypes, each depending on the clinical presentation.[7]
IBS is probably less common than BCIE, with less than 20 kindred reported in the English dermatological literature.[3] The mode of inheritance in most of the reported cases was autosomal-dominant though sporadic cases have been reported.[3],[8] Clinically, IBS is characterized by mild neonatal disease, or delayed onset with episodes of superficial blistering resembling epidermolysis bullosa simplex persisting through childhood, and sometimes into adult life. These blisters quickly rupture to produce annular peeling revealing superficially denuded skin, classically described as the mauserung phenomenon (moulting of the epidermis). Further, it is characterized by variable gray, rippled hyperkeratosis on the limbs and lower trunk with flexural accentuation. This hyperkeratosis is not as severe as in BCIE. Erythroderma does not occur in patients with IBS, although it has been described in one case report.[3] Other features described are palmoplantar blistering, hyperhidrosis, hypertrichosis, and pustulation.[2],[3]
Mechanical trauma, heat, and possibly profuse sweating play an important role in the formation of blisters, which heal without scarring or atrophy.[2] The flaccid blisters and mauserung appearance are apparently related to the histological phenomenon of superficial EHK, leading to reduced cohesion of keratinocytes. In EHK, abnormal keratin filament formation leads to an abnormal cytoskeleton, resulting in increased mechanical fragility. In addition, the desmosomal attachments are imperfect, which leads to blister formation. In contrast to BCIE and other diseases displaying EHK, the acanthokeratolysis is confined to the cells of the granular layer and the upper spinous layer in IBS. Electron microscopy shows granular cell edema with abundant keratin filament aggregates in the upper spinous layer. In the granular layer, the aggregates are associated with large keratohyaline granules.[9]
Recent studies have established a mutation of the gene for keratin 2 (K2e), the expression of which involves the third and fourth epidermal layers.[3],[8] The most commonly reported mutation, E493K, represents a mutational hotspot for IBS.[3]
Emollients and mild keratolytic preparations suffice as therapy in most, and often appear to induce an impressive remission.[9] Widespread bullous impetigo is a common complication in the early blistering phase and appropriate antibiotics are to be used when indicated. Furthermore, genetic counseling is an important part of the overall management of families with IBS.
Although the occurrence of similar disease in generations prior to one previous generation was not observed in our family of IBS, it may be presumed that the disease had occurred as a sporadic mutation in the previous generation (mother of our index patient), which in turn was carried onto our index patient. Both our patients share the clinical and histological features of the distinct and well-described entity of ichthyosis bullosa of Siemens. This applies in particular to the superficial blistering, the pattern of rippled hyperkeratosis of the limb flexures, and to the mauserung phenomenon. However, the superficial blistering occurred spontaneously rather than at trauma-prone sites and the mauserung phenomenon was observed extensively over the trunk and extremities, with striking resemblance to PSS, in contrast to the limited involvement observed in most reported cases of IBS.[1],[2],[3],[8],[10] This led us to ponder whether we had stumbled on a new variant of exfoliative ichthyosis sharing the cutaneous features of IBS and PSS. Zvulunov et al. has noted similar sharing of the cutaneous manifestations of IBS and PSS in a Bedouin family.[11] EHK was not, however, observed histologically in his patient. Histological demonstration of EHK in our patient supports a diagnosis of IBS. Our index patient probably belongs to the NPS-2 subtype of EHK classification proposed by DiGiovanna.[7] Unfortunately, electron-microscopic study or keratin study for a definitive diagnosis of IBS could not be done because these facilities were not available in our institution. But then, EHK is known to be a clinically heterogeneous disorder with variable disease severity and expression. This heterogeneity bears testimony to the clinical and histological diagnosis of IBS.
To conclude, IBS is a rare, distinct entity with characteristic cutaneous, histological, and ultrastructural features, which help to differentiate it from other bullous ichthyosiform syndromes. This case has been reported for its rarity and to re-emphasize the recognition of this heterogeneous, yet distinct, manifestation of epidermolytic hyperkeratosis.
| 1. |
Siemens HW. Dichtung und Wahreit �ber die "Ichthyosis bullosa," mit Bemerkungen zur Systematik der Epidermolysen. Arch Dermatol Syph 1937;175:590-608. [Quoted by: Traupe H, Kolde G, Hamm H, Happle R. Ichthyosis bullosa of Siemens: a unique type of epidermolytic hyperkeratosis. J Am Acad Dermatol 1986;14:1000-5.]
[Google Scholar]
|
| 2. |
Traupe H, Kolde G, Hamm H, Happle R. Ichthyosis bullosa of Siemens: a unique type of epidermolytic hyperkeratosis. J Am Acad Dermatol 1986;14:1000-5.
[Google Scholar]
|
| 3. |
Basarab T, Smith FJ, Jolliffe VM, McLean WH, Neill S, Rustin MH, et al . Ichthyosis bullosa of Siemens: report of a family with evidence of a keratin 2e mutation and a review of the literature. Br J Dermatol 1999;140:689-95.
[Google Scholar]
|
| 4. |
Dave S, Rakhesh SV, Thappa DM. Bullous congenital ichthyosiform erythroderma-PS1 type. Indian J Dermatol 2003;48:108-11.
[Google Scholar]
|
| 5. |
Brocq L. Erythrodermie congιnitale ichthyosiforme avec hyperepidermotrophie. Ann Dermatol Syphiligr 1902;4:1-31. [Quoted by: DiGiovanna JJ, Bale SJ. Clinical heterogeneity in epidermolytic hyperkeratosis. Arch Dermatol 1994;130:1026-35.]
[Google Scholar]
|
| 6. |
Traupe H. The epidermolytic (acanthokeratolytic) ichthyosis. In : The ichthyosis. New York: Springer-Verlag Inc; 1989. p. 139-53.
[Google Scholar]
|
| 7. |
DiGiovanna JJ, Bale SJ. Clinical heterogeneity in epidermolytic hyperkeratosis. Arch Dermatol 1994;130:1026-35.
[Google Scholar]
|
| 8. |
Kim SC, Hur W, Won JH, Ahn SK. Ichthyosis bullosa of Siemens: report of a sporadic case. J Dermatol 1995;22:283-8.
[Google Scholar]
|
| 9. |
Griffiths WA, Judge MR, Leight IM. Disorders of cornification. In : Champion RH, Burton JL, Burns DA, Breathnach SM, editors. Rook/Wilkinson/Ebling Textbook of Dermatology. 6th ed. Oxford: Blackwell Science; 1998. p. 1483-589.
[Google Scholar]
|
| 10. |
Thappa DM, Jeevankumar B. Ichthyosis Bullosa of Siemens. Indian Pediatr 2003;40:265,266.
[Google Scholar]
|
| 11. |
Zvulunov A, Cagnano E, Kachko L, Shorer Z, Elbedour K, Stevens H. A new variant of autosomal recessive exfoliative ichthyosis. Pediatr Dermatol 2002;19:382-7.
[Google Scholar]
|
Fulltext Views
3,343
PDF downloads
1,370





![[Figure - 1]](#fig_ijdvl_2006_72_1_43_19718_1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2006_72_1_43_19718_2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2006_72_1_43_19718_3.jpg){kind=link}
![[Figure - 4]](#fig_ijdvl_2006_72_1_43_19718_4.jpg){kind=link}