Translate this page into:
Tuberous sclerosis complex associated with dyschromatosis universalis hereditaria
Correspondence Address:
M P Binitha
'Haritha' (PO), Beypore, North Calicut - 673 015, Kerala
India
| How to cite this article: Binitha M P, Thomas D, Asha L K. Tuberous sclerosis complex associated with dyschromatosis universalis hereditaria. Indian J Dermatol Venereol Leprol 2006;72:300-302 |
Abstract
Tuberous sclerosis is an autosomal dominant disease due to mutations in two genetic loci, characterized by hamartoma formation in the skin, nervous system, heart, kidney, and other organs. Dyschromatosis universalis hereditaria is an autosomal dominant genodermatosis, characterized by small hyperpigmented and hypopigmented macules, uniformly distributed over the entire body. The face is rarely involved, and the palms, soles, and mucous membranes are usually spared. We report a case of tuberous sclerosis with dyschromatosis universalis hereditaria, with hyperpigmented and hypopigmented macules affecting the palms, soles, and oral mucosa. To our knowledge, this is the first reported case of such an association.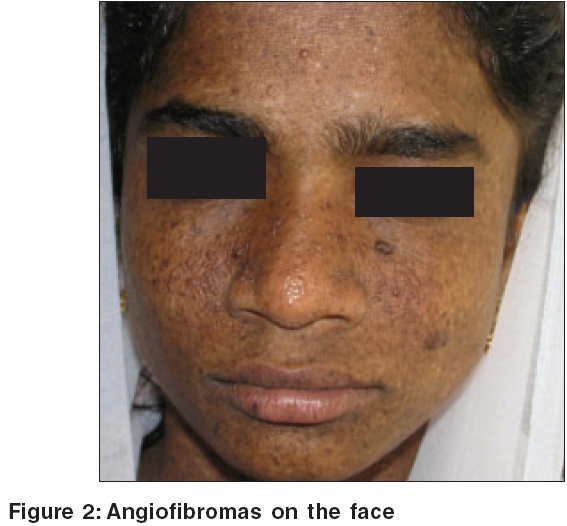 |
|
 |
|
Introduction
Tuberous sclerosis was first described by Von Recklinghausen in 1862. The name ′sclerosis tubereuse′ was used by Bourneville in 1880 to describe the firm consistency (sclerosis) of the protruberances (tuberosus). The term tuberous sclerosis complex (TSC) is currently used to describe the myriad manifestations due to hamartoma formation in multiple organ systems.[1] Dyschromatosis universalis hereditaria (DUH) was first reported by Jehikawa and Hiraga in 1933. It has been described almost exclusively in Asian families. Hereditary genetic defects may play an important role in altering melanogenesis, which results in pigmentary anomalies.[2] The reported associations include short stature, high tone deafness,[3] solar elastosis,[4] and grand mal epilepsy.[5]
Case report
A 17-year-old girl presented with progressively increasing, generalized, mottled pigmentation since birth. She also had a history of generalized epileptic seizures since the age of 3 years, mild global developmental delay, and a learning disorder. The patient′s family history was notable in that, 3 generations of her family had a similar pigmentary disorder including her sister, father, 5 out of 7 paternal uncles and aunts, and her paternal grandfather. However, there was no family history of other skin lesions or seizures.
On examination, the patient was of short stature with a height of 142 cm. There were multiple, randomly and uniformly distributed, small, irregular, hyperpigmented and hypopigmented macules without atrophy all over the body, including palms [Figure - 1], soles, and oral mucosa. She also had multiple angiofibromas on the face [Figure - 2], numerous oval, hypopigmented macules all over the trunk, confetti skin lesions, a shagreen patch over the right lumbosacral area, periungual fibromas on the left index finger and right third toe, gingival fibromas, and pitting of dental enamel.
Histopathological examination from the hyperpigmented and hypopigmented macules revealed increased melanin content, and decreased melanin content in the epidermis, respectively. The melanocyte number was normal in both specimens. Neurodevelopmental testing demonstrated mild mental retardation and learning disability. Electroencephalography was normal. Computed tomography of the head demonstrated periventricular subependymal calcified tubers. Magnetic resonance imaging examination of the head and brain was normal. Echocardiography showed hyperechoic shadows in the interventricular septum, characteristic of regressing rhabdomyomas. Ophthalmological examination, renal ultrasonography, skeletal radiographs of skull, chest, hands and feet, electrocardiograph, and pulmonary function tests were within normal limits. Genetic studies could not be done due to lack of facilities.
Examination of the family members revealed generalized hyperpigmented and hypopigmented macules affecting the patient′s father, sister, and paternal grandfather. There was no mucosal or palmoplantar involvement. They did not show clinical features of TSC. Ophthalmological examination and renal ultrasonography were normal in both parents.
Discussion
The revised diagnostic clinical criteria for TSC have been divided into major and minor criteria.[6]
Major features 1. Facial angiofibromas or forehead plaque
2. Nontraumatic ungual or periungual fibromas
3. Hypomelanotic macules (>3)
4. Shagreen patch (connective tissue nevus)
5. Multiple retinal nodular hamartomas
6. Cortical tuber
7. Subependymal nodule
8. Subependymal giant cell astrocytoma
9. Cardiac rhabdomyoma, single or multiple
10. Lymphangiomyomatosis
11. Renal angiomyolipoma
Minor features 1. Multiple randomly distributed pits in dental enamel
2. Hamartomatous rectal polyps
3. Bone cysts
4. Cerebral white matter radial migration lines
5. Gingival fibromas
6. Nonrenal hamartoma
7. Retinal achromic patch
8. Confetti skin lesions
9. Multiple renal cysts
Definite tuberous sclerosis complex is diagnosed by the presence of either 2 major features, or 1 major feature plus 2 minor features. Probable tuberous sclerosis complex is indicated by 1 major feature plus 1 minor feature. Possible tuberous sclerosis complex is indicated by either 1 major feature, or 2 or more minor features. Our case had 6 major and 3 minor criteria, diagnostic of definite TSC, along with definite features of DUH, which was also present in 3 generations in an autosomal dominant pattern. However, there was no evidence of TSC in her parents or siblings.
Two genetic loci have been identified in TSC. The first gene, tuberous sclerosis complex 1 (TSC 1), maps to chromosome 9, specifically 9q34, and encodes the protein hamartin, which is a tumor suppressor gene. The second gene (TSC 2) maps to chromosome 16, specifically 16p13, and codes for tuberin. TSC 2 acts as a tumor-suppressor gene, and TSC 2 mutations may result in tumors at multiple sites. Hamartin and tuberin act synergistically to regulate cellular growth and differentiation. The deregulation in organogenesis results in hamartomas, which may affect any organ in the body.[7]
Dyschromatoses are a group of disorders characterized by the presence of both hyperpigmented and hypopigmented, small, irregular macules. DUH is an autosomal dominant disorder of melanosome synthesis rate or melanosome activity, and not a disorder of melanocyte number. The gene responsible for DUH has been mapped to 6q24.2 to 6q25.2 (OMIM 127500). Since the exact biochemical basis of the gene defect is unknown, the clinician must rely solely on external phenotype. DUH has been reported in association with another rare autosomally inherited pigmentary disorder, Dowling-Degos disease,[8] and also with X-linked ocular albinism.[9] The exact mechanism by which these genetic disorders have occurred simultaneously, has not been elucidated. Autosomal dominant traits can involve only one organ as in DUH. However, many autosomal disorders like TSC can manifest in a number of different systems of the body, called pleiotropy.
To the best of our knowledge, there are no previous reports of TSC with DUH, or any other genetic disorder. Although it is possible that our patient had two genetic disorders due to sheer chance, it is important to record any such association, since it may indicate genetic linkage. The unusual features in our case were the involvement of palms, soles, and oral mucosae.
| 1. |
Harper JI. Genetics and genodermatoses. In : Champion RH, Burton JL, Burns DA, Breathnach SM, editors. Rook / Wilkinson / Ebling Textbook of Dermatology. 6th ed. Oxford: Blackwell Science; 1998. p. 357-436.
[Google Scholar]
|
| 2. |
Sethuraman G, Srinivas CR, D'Souza M, Thappa DM, Smiles L. Dyschromatosis universalis hereditaria. Clin Exp Dermatol 2002;6:477-9.
[Google Scholar]
|
| 3. |
Rycroft RJ, Colman CD, Wells RS. Universal dyschromatosis, small stature and high tone deafness. Clin Exp Dermatol 1977;2:45-8.
[Google Scholar]
|
| 4. |
Sait MA, Garg BR. Dyschromatosis universalis hereditaria. Indian J Dermatol Venereol Leprol 1985;51:277-9.
[Google Scholar]
|
| 5. |
Pavithran K. Dyschromatosis universalis hereditaria with epilepsy. Indian J Dermatol Venereol Leprol 1991;57:102-3.
[Google Scholar]
|
| 6. |
Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol 1998;12:624-8.
[Google Scholar]
|
| 7. |
Tuberous sclerosis. http://www.emedicine.com/DERM/topic438.htm. Last accessed: 26th December 2005.
[Google Scholar]
|
| 8. |
Sandhu K, Saraswat A, Kanwar AJ. Dowling-Degos' disease with dyschromatosis universalis hereditaria-like pigmentation in a family. J Eur Acad Dermatol Venereol 2004;18:702-4.
[Google Scholar]
|
| 9. |
Yang JH, Wong CK. Dyschromatosis universalis with X-linked ocular albinism. Clin Exp Dermatol 1991;16:436-40.
[Google Scholar]
|
Fulltext Views
3,184
PDF downloads
2,757





![[Figure - 1]](#fig_ijdvl_2006_72_4_300_26729_1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2006_72_4_300_26729_2.jpg){kind=link}