
Translate this page into:
Approach to inherited hypertrichosis: A brief review
Corresponding author: Dr. Jeta Buch, E-13, Basement, Defence Colony, Near Andrews Ganj Bus Stop, Delhi, India. jetabuch@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Buch J, Ranganath P. Approach to inherited hypertrichosis: A brief review. Indian J Dermatol Venereol Leprol 2022;88:11-21.
Abstract
Hypertrichosis refers to the growth of hair, of an excessive amount and thickness, on any part of the body. It must be distinguished from hirsutism which is characterized by excess growth of hair in androgen-dependent areas on the upper lip, chin, chest, linea alba, thigh and axilla. Hypertrichosis may be localized or generalized, and congenital or acquired. Excess hair growth has a psychological impact on the child as well as the parents due to the cosmetic disfigurement it produces. Current treatment options are limited and not wholly satisfactory. Treatment should be customized according to the area, nature and amount of hair growth, age of the patient and personal preferences. In addition, when hypertrichosis occurs as a component of a syndrome, multidisciplinary management is required to address the associated systemic features. A detailed review of inherited generalized hypertrichosis is presented here with emphasis on clinical clues to identifying complex syndromes with multisystem involvement.
Keywords
Generalized
hypertrichosis
inherited

Introduction
The normal sequence of development of hair form and pattern varies widely in childhood and adolescence. One of the major categories of abnormal hair growth patterning is hypertrichosis, defined as the growth of hair (lanugo/vellus/ terminal) on any part of the body in excess of the amount that is normally present in persons of the same age, race and sex and excluding androgen-induced patterns of hair growth.1 It is usually a cosmetic problem, but in children seldom persists as an isolated symptom for long, becoming linked to alterations in somatic growth, sexual function or general metabolism, often forming a component of major malformation syndromes.2
Methods
A literature search of scientific publications published in English was done using the electronic databases PubMed (https://pubmed.ncbi.nlm.nih.gov/), GeneReviews (https://www.ncbi.nlm.nih.gov/books/NBK1116/), OMIM (https://omim.org/) and Google Scholar (https://scholar.google.co.in/). Search included terms “hypertrichosis,” “congenital,” “acquired,” “localized,” “generalized” and “treatment.” One hundred forty nine articles and references within the articles so obtained were reviewed to identify additional studies available. Studies and case reports which met the following criteria were included in the current review:
English language publications
Those focusing exclusively on inherited hypertrichosis.
Data obtained from studies and case reports were compiled and interpreted to prepare this review.
Results
Hypertrichosis should be differentiated from hirsutism, which is increased hair growth in androgen-dependent areas. Unexplained hirsutism in childhood usually warrants investigation to exclude an endocrine cause for the virilization.3
Hypertrichosis is classified as generalized and localized with further subdivision into congenital and acquired forms. Hypertrichosis may either be an isolated finding or be associated with other abnormalities [Figures 1-3].
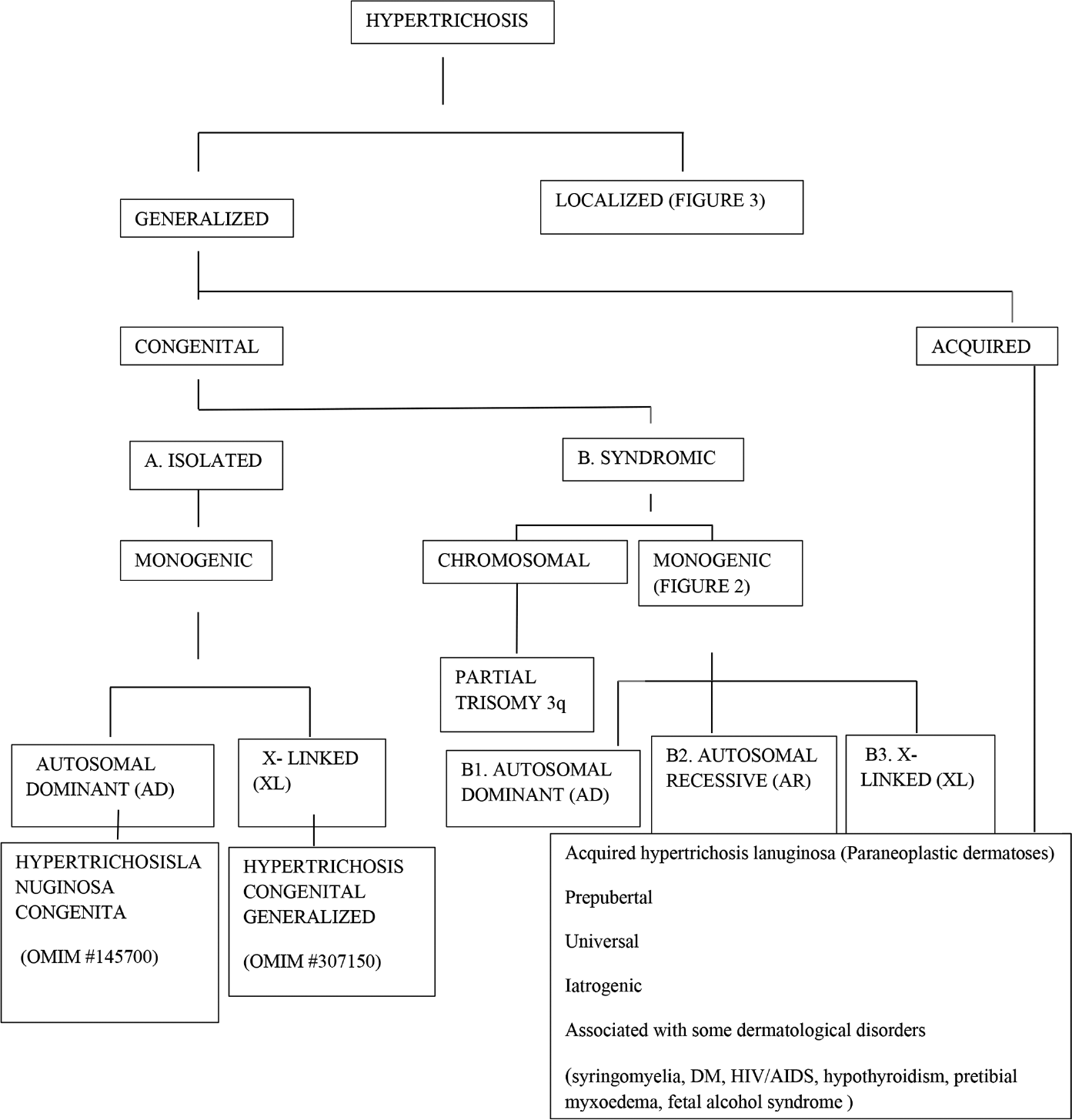
- Classification of hypertrichosis
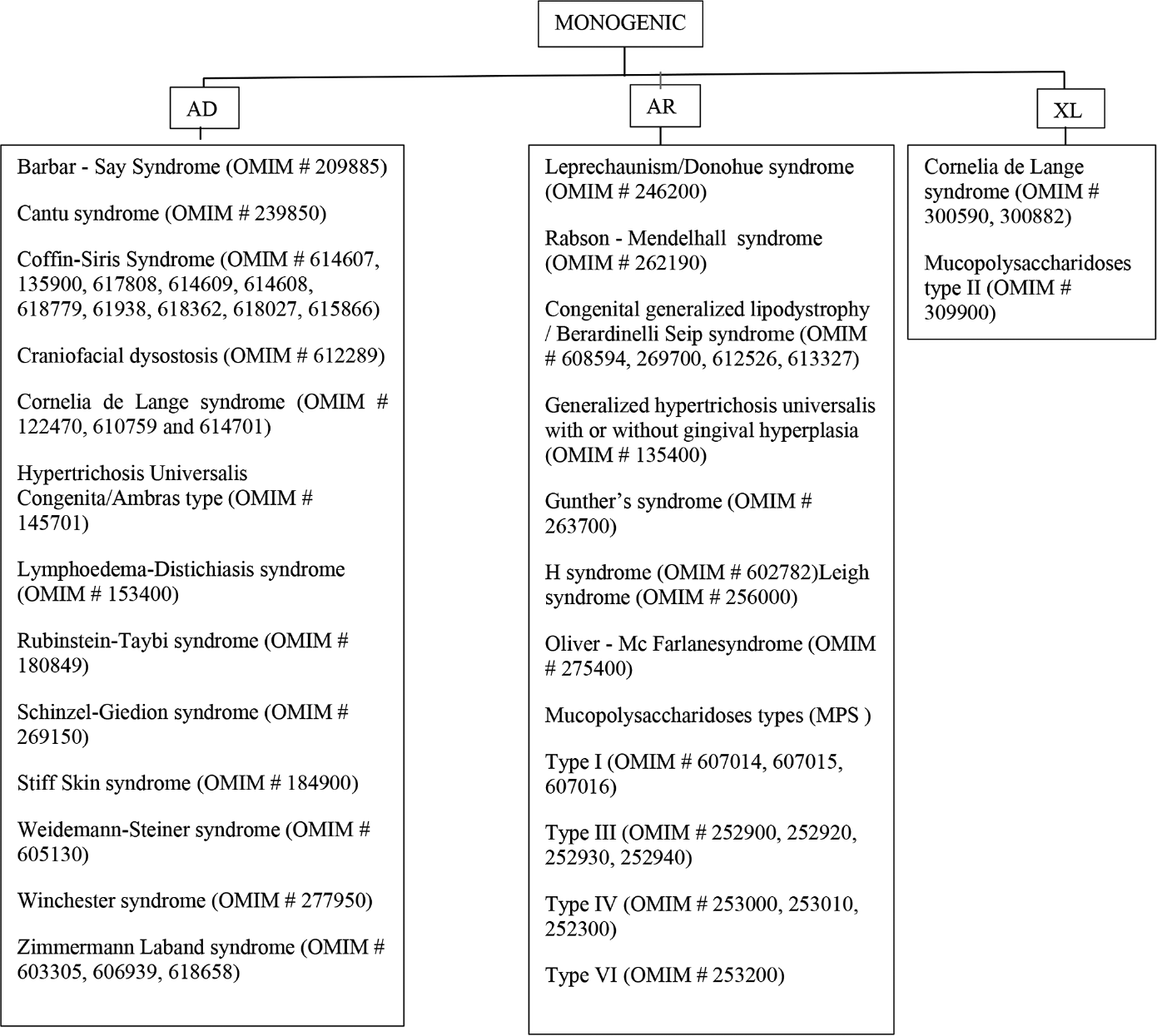
- Monogenic syndromic congenital generalized hypertrichosis
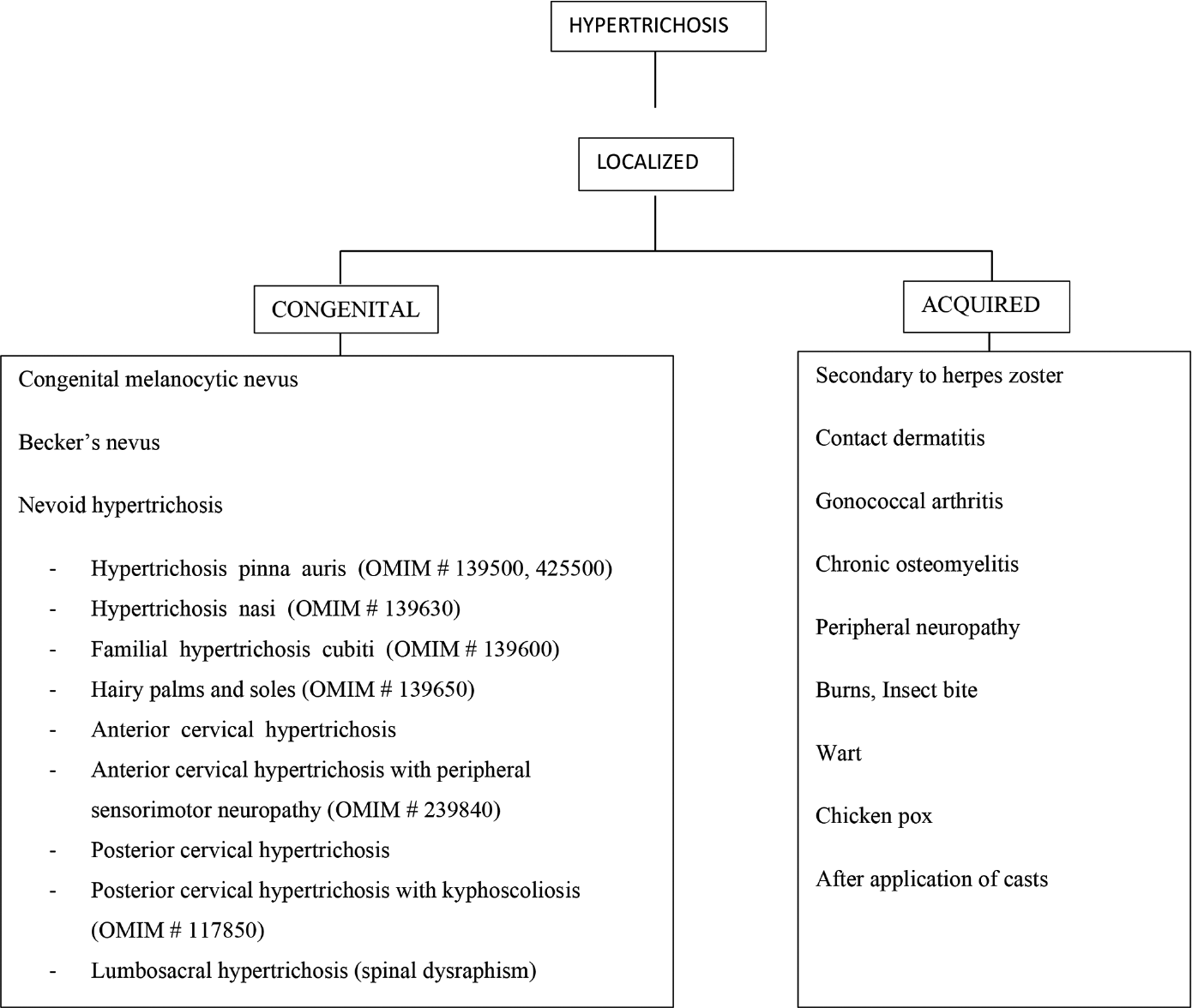
- Classification of localized hypertrichosis
Clinical Features
A. Isolated congenital hypertrichosis
Monogenic
-
Hypertrichosis lanuginosa congenita
Inheritance/gene locus: autosomal dominant/not known
Clinical features: congenital hypertrichosis lanuginosa is a rare disorder characterized by excessive lanugo hair from birth, homogeneously covering the entire body except the mucosae, palms and soles.4 During the first year of life, hair shedding begins on the trunk and progresses to the arms and legs.
-
Hypertrichosis congenita terminalis
Inheritance/gene locus: X-linked dominant/ palindrome-mediated inter-chromosomal insertion on chromosome Xq27.1
Clinical features: in hypertrichosis congenita terminalis, males are more frequently affected than females. Affected males have a generalized distribution of terminal hair, whereas hypertrichosis in females is asymmetric and patchy, consistent with lyonization.5
B. Syndromic congenital hypertrichosis
Chromosomal
Partial trisomy 3q
Clinical features: partial trisomy 3q is a rarely reported chromosomal disorder. Duplication of a part of the long arm of chromosome three leads to hypertrichosis, typical craniofacial dysmorphism (prominent forehead, epicanthic fold, broad and flat nasal bridge, anteverted nostrils, prominent philtrum, micrognathia, cleft lip and cleft palate, downturned corners of the mouth and thin lips, malformed ears and short neck), cystic hygroma, brain malformations (microcephaly and Dandy-Walker malformation), ocular malformations (strabismus, nystagmus, corneal opacities, cataract, colobomas of the iris, coloboma of optic nerve and anophthalmia), congenital heart defects (septal defects), renal malformations (polycystic kidneys or dysplasia), skeletal anomalies (hypoplasia of the phalanges, camptodactyly and clinodactyly) as well as a simian crease6
Monogenic conditions
Autosomal dominant disorders
Barbar-Say syndrome
Inheritance/gene locus: autosomal dominant/TWIST2
Clinical features: Barber-Say syndrome is a rare congenital disorder characterized by severe hypertrichosis mainly over the back, sparse eyebrows and eyelashes, redundant, generalized, hyperlax and atrophic skin and facial dysmorphism including low frontal hairline, ocular telecanthus, hypoplastic eyelids, periorbital fullness, ectropion, broad nasal bridge, bulbous nose, hypoplastic alae nasi, thick-flared alae nasi, anteverted nares, cheek pads, macrostomia, thin upper vermilion border, micrognathia, low-set posteriorly rotated ears, small/ absent mammary glands and small/absent nipples.7
Cantu syndrome
Inheritance/gene locus: autosomal dominant/ABCC9
Clinical features: Cantu syndrome, also known as hypertrichotic osteochondrodysplasia, is characterized by congenital generalized hypertrichosis, lymphedema, fetal finger pads, soft “dough-like” skin, deep-set nails, macrocephaly, coarse facies, hypertelorism, long palpebral fissures, epicanthal folds, flat nasal bridge, long philtrum, wide mouth, thick lips and mild intellectual disability. Cardiac manifestations include patent ductus arteriosus, septal hypertrophy, pulmonary hypertension, pericardial effusion, and skeletal abnormalities include thickened calvarium, narrow thorax, wide ribs, flattened or ovoid vertebral bodies, coxa valga, osteopenia, enlarged medullary canals and metaphyseal widening of long bones.8
Coffin-Siris syndrome
Inheritance/gene locus: autosomal dominant/ ARID1A, ARID1B, ARID2, SMARCA4, SMARCB1, SMARCD1, SMARCE1, SMARCE2, DPF2 and SOX11
Clinical features: typical features of Coffin-Siris syndrome include generalized hypertrichosis with scalp hypotrichosis, absence of fifth fingernails and toenails, absence of distal phalanges of fifth fingers and of the second to fifth toes, inguinal hernia, sucking and feeding difficulty, recurrent upper and lower respiratory tract infections, decreased fetal activity and intrauterine growth retardation. Other features associated with this syndrome are microcephaly, bushy eyebrows, long eyelashes, low, flat nasal bridge, wide upturned nasal tip, short philtrum, wide mouth, prominent lips and cleft palate. Postnatal growth retardation and moderate developmental delay are regular features in older children and adults.9
Craniofacial dysostosis-hypertrichosis/Fontaine progeroid syndrome/Gorlin-Chaudhry-Moss syndrome
Inheritance/locus: autosomal dominant/SLC25A24
Clinical features: craniofacial dysostosishypertrichosis syndrome is characterized by marked generalized hypertrichosis, pre- and post-natal growth retardation, cranial malformation (craniosynostosis), facial malformation (low scalp hairline, wide and open anterior fontanelle, triangular face, midfacial hypoplasia, small eyes, downslanting palpebral fissures, convex and broad nasal bridge), high arched and narrow palate, dental malocclusion, abnormally shaped teeth, oligodontia, microdontia, micrognathia, genital hypoplasia and persistent patent ductus.10
Cornelia de Lange syndrome
Inheritance/locus: autosomal dominant/NIPBL, SMC3, RAD21. X-linked dominant/SMC1A and HDAC8
Clinical features: the Cornelia de Lange syndrome11 phenotype can be described as a spectrum associated with abnormalities affecting genes that have a structural and regulatory function in the cohesin complex.
Cardinal features include (two points each if present)-
-
Facial [Figure 4]
Synophrys (meeting of the medial eyebrows in the midline) and/or thick eyebrows
Short nose, concave nasal ridge and upturned nasal tip
Long and/or smooth philtrum
Thin upper lip vermilion and/or downturned corners of the mouth.
-
Non-facial
Hand oligodactyly (the congenital absence of one or more fingers) and/or adactyly (the absence of all fingers and/or toes)
Congenital diaphragmatic hernia.
Suggestive features include (one point each if present)
Global developmental delay and/or intellectual disability
Prenatal growth retardation
Postnatal growth retardation
Microcephaly (prenatally and/or postnatally)
Small hands and/or feet
Short fifth finger
Hirsutism.
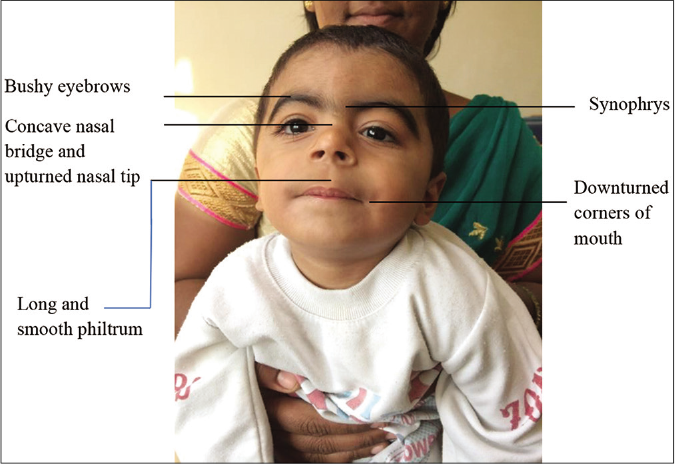
- Cardinal features of Cornelia de Lange syndrome (as shown)
The International Cornelia de Lange syndrome consensus group developed consensus criteria using these features — A score of -≥11 — classic Cornelia de Lange syndrome if at least three cardinal features are present regardless of whether a pathogenic variant in one of the known genes can be found.
9–10 — non-classic Cornelia de Lange syndrome if at least two cardinal features are present
≥4 — sufficient to warrant molecular testing if at least one cardinal feature is present
<4 — insufficient to indicate such testing.
Hypertrichosis universalis congenita/Ambras type
Inheritance/gene locus: autosomal dominant/complex rearrangement on chromosome 8/possible involvement of TRPS1
Clinical features: this condition is characterized by persistent generalized excessive vellus hair growth with accentuation of hypertrichosis over the face, ears and shoulders and sparing of palms, soles and mucosa. The forehead, eyelids, cheek, nose and preauricular region are uniformly covered with hair, which reaches a length of few centimeters.12 Other features include facial dysmorphism (triangular coarse face, bushy eyebrows, long eyelashes, telecanthus, long palpebral fissure, depressed nasal bridge, bulbous nose, rounded nasal tip, anteverted nares, thick nasal wings, long prominent philtrum, thin vermilion border and trapezoid mouth), skeletal abnormalities (post-axial rudimentary hexadactyly, exostosis and low insertion of the first metacarpal) and accessory nipples and teeth abnormalities (oligodontia, delayed primary/secondary dentition).
Lymphoedema-distichiasis syndrome
Inheritance/gene locus: autosomal dominant/FOXC2
Clinical features: Lymphoedema-distichiasis syndrome is characterized by lower limb lymphedema, which typically appears in late childhood or puberty, and is often asymmetric. Distichiasis (aberrant eyelashes arising from the meibomian glands), which may be present at birth, is observed in 94% of affected individuals. About 75% of individuals have corneal irritation, recurrent conjunctivitis and photophobia. Other common findings include varicose veins, congenital heart disease and ptosis.13
Rubinstein-Taybi syndrome
Inheritance/gene locus: autosomal dominant/CREBBP, EP300
Clinical features: Rubinstein-Taybi syndrome is characterized by intellectual disability, postnatal growth deficiency, striking dysmorphic features of the face (microcephaly, high-arched eyebrows, long eyelashes, downslanting palpebral fissure, broad nasal bridge, beaked nose, overhanging columella, high arched palate, grimacing and micrognathia) [Figure 5], hands and feet (broad thumb and hallux). Ocular anomalies such as nasolacrimal duct obstruction, ptosis, congenital or juvenile glaucoma and refractive errors and orodental features such as talon cusps, dental crowding, crossbite and screwdriver incisors are infrequent. Rubinstein-Taybi syndrome patients are prone to develop keloids and tumors of neural or developmental origin.14
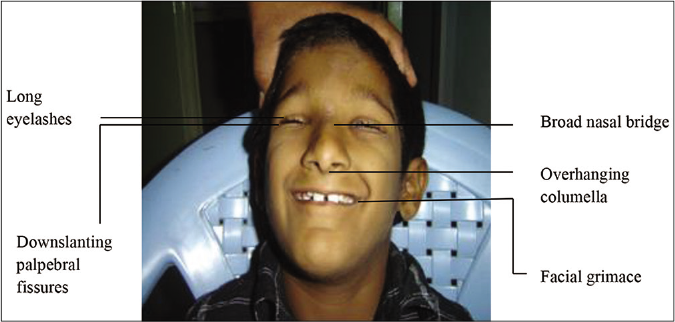
- Facial features of Rubinstein-Taybi syndrome
Schinzel-Giedion syndrome
Inheritance/gene locus: autosomal dominant/SETBP1
Clinical features: diagnosis of Schinzel-Giedion syndrome is based on clinical findings and/or SETBP1 mutation. Type 1 (complex and classic type) presents with developmental delay and typical facial features (prominent forehead, midface retraction, short and upturned nose) associated with hydronephrosis or two of the characteristic skeletal anomalies (sclerotic skull base, broad ribs, increased cortical density and wide occipital synchondrosis). Type 2 (middle type) shows developmental delay, distinct facial phenotype but lacks hydronephrosis and skeletal anomalies with the existence of SETBP1 mutation. Type 3 (simple type) with SETBP1 mutation presents with developmental delay in which expressive language delay is the striking feature.15 Hypertrichosis may often be the presenting feature that disappears in infancy.
Stiff skin syndrome
Inheritance/gene locus: autosomal dominant/FBN1
Clinical features: Stiff skin syndrome presents with rock-hard induration of skin over buttocks and thighs, with hypertrichosis and hyperpigmentation of overlying skin. Extracutaneous features include large joint contractures, scoliosis, tiptoe gait, narrow thorax, growth retardation and restrictive lung disease.16
Weidemann-Steiner syndrome
Inheritance/gene locus: autosomal dominant/SLC25A24
Clinical features: Weidemann-Steiner syndrome is associated with dysmorphic facies (thick eyebrows, long eyelashes, downslanting palpebral fissures, wide nasal bridge, broad nasal tip, thin upper lip and micrognathia), hypertrichosis, short stature, developmental delay and intellectual disability. Congenital malformations of the cerebral, cardiac, renal and optic structures have also been reported.17
Zimmermann-Laband syndrome
Inheritance/gene locus: autosomal dominant/KCNH1, ATP6V1B2 and KCNN3
Clinical features: the main clinical features of Zimmermann-Laband syndrome are gingival enlargement, bulbous soft nose, thick floppy ears, nail aplasia or hypoplasia, hypertrichosis, joint hyperextensibility, hepatosplenomegaly and intellectual disability with or without epilepsy.18
Autosomal recessive disorders
Leprechaunism/Donohue syndrome and RabsonMendenhall syndrome
Inheritance/gene locus: autosomal recessive/INSR
Clinical features: mutations in the insulin receptor gene INSR cause a spectrum of inherited insulin resistance syndromes: leprechaunism presenting the most severe phenotype with death with one year of life; Rabson-Mendenhall syndrome, an intermediate phenotype with survival beyond one year but death usually before puberty and type A insulin resistance, the milder form usually evident after puberty. Both leprechaunism and Rabson-Mendenhall syndrome have similar phenotypic manifestations characterized by distinct craniofacial features (small elfin face, large low-set ears, prominent eyes, wide nostrils, thick lips and largemouth), hypertrichosis, acanthosis nigricans, lipoatrophy, prenatal and postnatal growth retardation, precocious puberty, breast hyperplasia, prominent nipples, distended abdomen, large hands and feet, muscle atrophy, penile enlargement, clitoromegaly and polycystic ovaries. However, Rabson-Mendenhall syndrome differs from leprechaunism being less severe, with the presence of premature and dysplastic dentition, coarse facies, gingival hyperplasia, pineal hyperplasia and survival beyond one year of age.19-24
Congenital generalized lipodystrophy/Berardinelli-Seip syndrome
Inheritance/gene locus: autosomal recessive/AGPAT2, BSCL2, CAV1 and PTRF
Clinical features: this syndrome is characterized by signs and symptoms of insulin resistance such as hypertriglyceridemia, muscle hypertrophy, hepatic steatosis (which may progress to cirrhosis and death due to hepatic failure), diabetes mellitus, acute pancreatitis, premature cardiovascular disease, acanthosis nigricans and increase in appetite (due to low serum levels of adipocytokines such as leptin and adiponectin). An acromegaloid appearance [Figure 6], with minor enlargement of the mandible, hands and feet due to insulin-like growth factor 1 hypersecretion is seen during childhood. In adolescence, insulin resistance increases the risk of the polycystic ovarian syndrome in females whereas males have normal fertility.25
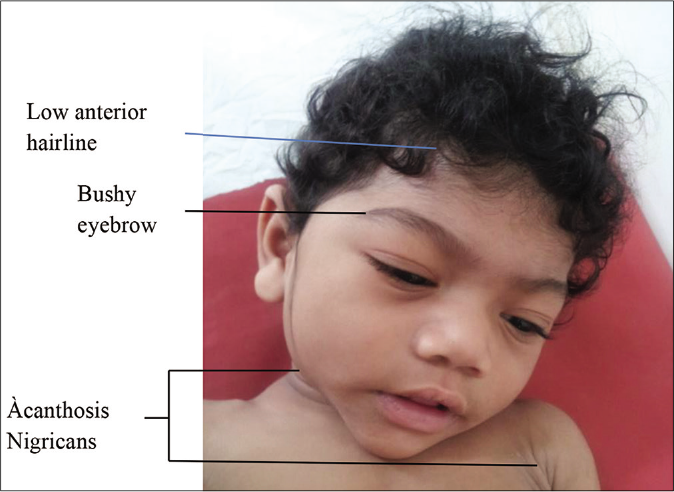
- Facial features of Berardinelli-Seip syndrome include mild coarsening, acromegaloid appearance, low anterior hairline, bushy eyebrows, large ears
Congenital erythropoietic porphyria (Gunther disease)
Inheritance/gene locus: autosomal recessive/UROS
Clinical features: the disease is characterized by early photosensitivity leads to erythema, blisters and erosions that leave scars. Secondary infection with bone resorption may lead to deformity and disfigurement of fingers, toes and facial features including nose, ears and eyelids. Focal hypo or hyperpigmentation and hypertrichosis of extremities may occur. Porphyrin deposition in the enamel and dentine of teeth results in “erythrodontia” imparting a dirty yellow or brown color in sunlight, and a brilliant red color under Wood’s lamp, and red urine, bone loss, ectropion, keratoconjunctivitis, cataract and blindness. All patients present with hemolytic anemia and splenomegaly.26
Generalized hypertrichosis universalis with or without gingival hyperplasia
Inheritance/locus: autosomal recessive/ABCA5
Clinical features: this condition is characterized by congenital generalized hypertrichosis at birth or during early infancy, but in up to half of the cases, it begins during puberty, manifesting mainly on the face, upper limbs and middle of the back. Gingival hyperplasia is almost always seen after the age of 10. Dysmorphic features such as downslanting palpebral fissures, epicanthal fold, bulbous tip of nose, thick alae nasi, broad root of nose and long prominent or deep philtrum have been documented in occasional patients.
Histiocytosis lymphadenopathy plus syndrome (H syndrome)
Inheritance/gene locus: autosomal recessive/SLC29A3
Clinical features: the cardinal cutaneous features of H syndrome include progressive symmetric cutaneous induration with hyperpigmentation and hypertrichosis localized mainly to the lower limbs [Figure 7] and lower abdomen. Cardinal extracutaneous features include hepatosplenomegaly, sensorineural hearing loss, hyperglycemia (nonautoimmune diabetes mellitus), heart anomalies, hypogonadotropic hypogonadism, growth hormone deficiency manifesting as short stature and hallux valgus/flexion contractures. Male subjects have been reported to have gynecomastia, scrotal masses and azoospermia.27,28
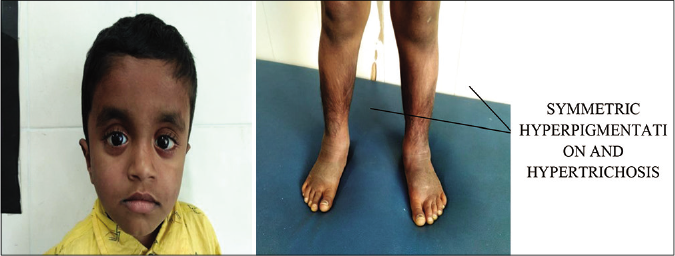
- Cutaneous features of H syndrome
Leigh syndrome
Inheritance/gene locus: autosomal recessive/SURF1
Clinical features: Leigh syndrome manifests in the central nervous system as progressive psychomotor retardation, nystagmus, ophthalmoparesis, optic atrophy, ataxia, dystonia and respiratory failure. Some patients present with peripheral nervous system involvement, including polyneuropathy or myopathy, non-neurologic abnormalities and hypertrichosis.29
Oliver-McFarlane syndrome
Inheritance/gene locus: autosomal recessive/PNPLA6
Clinical features: also termed as chorioretinopathy pituitary dysfunction syndrome, it is characterized by physical and intellectual disability as a result of untreated thyroid and growth hormone abnormalities, along with retinitis pigmentosa and trichomegaly. Congenital hypogonadism occurs in half of the patients, and nearly all have documented hypogonadotropic hypogonadism during puberty, with subsequent reproductive dysfunction.30
Mucopolysaccharidoses
Inheritance: autosomal recessive (except mucopolysaccharidoses II or Hunter syndrome which has an x-linked recessive pattern)
Clinical features: seven types are currently known based on the underlying mutation and consequent degree of enzyme activity, age of onset (earlier onset having a more severe disease), systemic involvement and clinical course.31,32
Mucopolysaccharidoses 1 has three main subtypes, all caused by a deficiency of the lysosomal enzyme alpha-L-iduronidase, due to biallelic mutations in the IDUA gene:
Mucopolysaccharidoses-1H or Hurler syndrome is characterized by a severe phenotype including coarse facies with thickened and dry skin, hypertrichosis on the face, trunk and extremities, Mongolian spots, intellectual disability, neurological deficits, hepatosplenomegaly, joint contractures, dysostoses multiplex, short stature, corneal clouding, cardiac valvular disease, hydrocephalus and hernias [Figure 8]
Mucopolysaccharidoses 1S or Scheie syndrome has a milder phenotype with normal intellectual capabilities and normal stature, with survival until adulthood
Mucopolysaccharidoses 1H/S is the intermediate phenotype with macrocephaly, macroglossia and joint contractures, with normal intellect or mild intellectual disability.
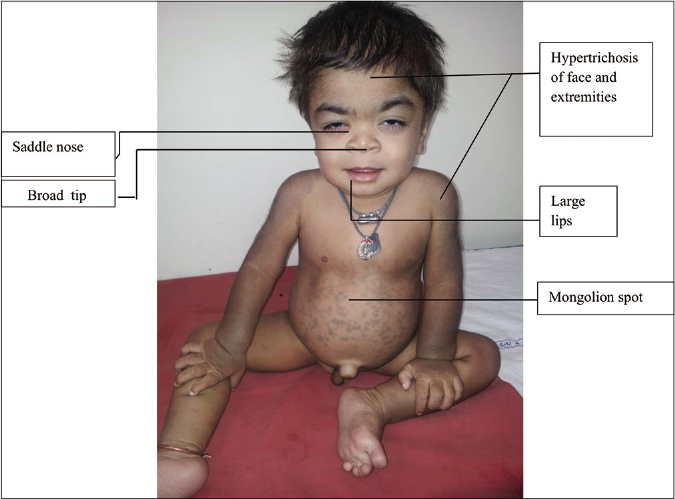
- Features of Mucopolysaccharidosis -1H (Hurler syndrome)
Mucopolysaccharidoses II or Hunter syndrome [Figure 9] has two forms. One is the less severe, mucopolysaccharidoses 1 Hunter syndrome-like form which manifests late with pigmentary changes on fundoscopy along with a loss of visual acuity, sclerodermoid infiltration of extremities, multiple ivory-white papules in the subscapular area and hypertrichosis. The other is the mucopolysaccharidoses 1H-like condition, which, in addition to the above, presents with hydrocephalus, hoarseness, respiratory difficulty, diarrhea and degenerative arthritis. Progressive neurological deterioration leads to death. The hypertrichosis appears from two to four years of age.32
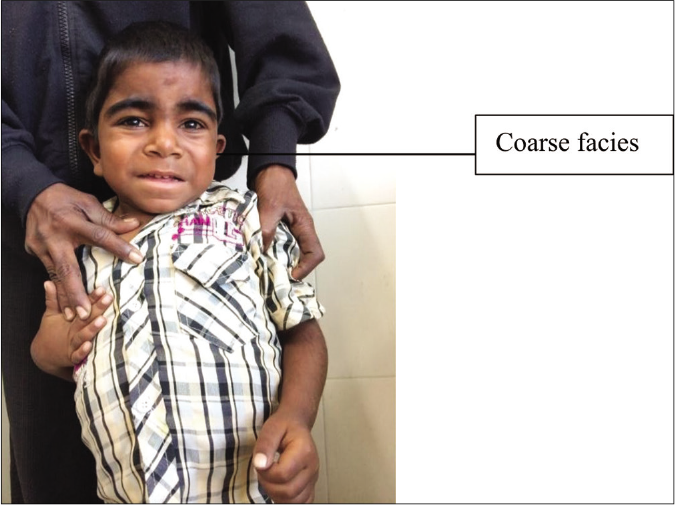
- Cardinal features of mucopolysaccharidosis II include male preponderance, hypertrichosis, coarse facies, hepatosplenomegaly, sclerodermoid infiltration of extremities, multiple ivory white papules in subscapular area
Mucopolysaccharidoses III or Sanfilippo syndrome [Figure 10] is characterized by hypertrichosis with severe intellectual disability, behavioral disorders, aggressive behavior and survival until the second or third decades. Mucopolysaccharidoses IV or Morquio syndrome is associated with short stature, corneal haziness, cardiac disease, and spondyloepiphyseal skeletal dysplasia with pectus carinatum, lumbar kyphosis and genu valgus deformity. Mucopolysaccharidoses VI or Maroteaux-Lamy syndrome is associated with short stature from the age of two–three years, coarse facies, corneal clouding and hepatosplenomegaly, with normal intellect.32

- Bushy eyebrows in mucopolysaccharidosis III

- Hypertrichosis in mucopolysaccharidosis III
Winchester syndrome
Inheritance/gene locus: autosomal recessive/MMP14
Clinical features: Winchester syndrome is characterized by skeletal radiologic characteristics (multifocal osteoporosis, progressive osteolysis and degenerative changes in the vertebral bodies, carpal and tarsal bones, including the metaphyses and epiphyses) with at least two of the following features: short stature and progressive articular contractures, corneal opacities, leathery skin or hypertrichosis, hypertrophy of the gums and coarsened facial features.33
Genetic Evaluation
Molecular genetic testing is now the preferred modality for diagnosis and characterization of the exact genetic etiology associated with hypertrichosis. For a few conditions, other confirmatory tests might be available, for example, lysosomal enzyme assays for the mucopolysaccharidoses. The availability of next-generation sequencing has significantly reduced the cost and time for sequence analysis of the different genes associated with hypertrichosis. Next-generation sequencing has also vastly improved the mutation detection rate and helped in the identification of many novel genes and gene variants related to hypertrichosis. If the clinical diagnosis suggests the possibility of involvement of a specific gene that has only a small number of exons, Sanger sequencing of that gene alone might be done. But for conditions that are genetically heterogeneous or with overlapping phenotypes or with large genes, a next-generation sequencing-based focused exome sequencing panel can be used as the first-tier test. If no significant variants are found, whole-exome sequencing and/or whole-genome sequencing can be done further.
Following an accurate diagnosis of the affected individual, appropriate genetic counseling can be provided to the patient and the family about the nature of the disorder, natural course and prognosis, available management options and the surveillance/monitoring plan. Many of the hypertrichosis-associated genetic syndromes have associated systemic/ multiorgan involvement, as mentioned above, and therefore require multidisciplinary management.
Based on the identified genetic etiology, the pattern of inheritance, that is, autosomal dominant, autosomal recessive or X-linked can be ascertained and the risk of recurrence in subsequent pregnancies in the family can be determined. Carrier testing of other family members is also possible. Prenatal diagnosis can be offered for at-risk pregnancies through targeted mutation analysis after identifying the pathogenic variant(s) in the affected proband and/or carrier parents, to prevent the recurrence of severe genetic disorders in the family.
Treatment
The necessity for treatment depends on the age of the patient, site and degree of hypertrichosis as also the psychosocial impact. Embarrassment, feelings of alienation experienced by parents in an excessively hairy newborn or child, and the need to keep a profusely hairy nappy area clean may call for extreme measures such as neonatal shaving.
Different methods of hair removal in generalized hypertrichosis include shaving (conventional/electric), trimming, bleaching, waxing, chemical depilatories, electrolysis and laser hair removal.
Shaving
Conventional shaving can damage the stratum corneum which, in turn, can stimulate nerve endings and activate cell signaling pathways that trigger the release of cytokines. These cytokines may cause burning, itch and increased blood perfusion resulting in erythema. A compromised stratum corneum lipid barrier may cause transepidermal water loss, more so in neonates due to their high body surface area: volume ratio, increased blood flow and thin epidermis leading to dryness and itch.
In electric shaving, interaction of the stratum corneum with the shaver’s cutter when pressed into the small apertures of shaving foil results in irritation. Modern electric shavers have inbuilt specialized trimmers in direct association with the apertured foil designed to tackle longer hair and lowlying hair that grow in multiple directions; vibration of the shaver foil or cutting elements at an ultrasonic rate is claimed to increase the uptake of hairs. These features may act synergistically to improve shaving efficiency, thoroughness, closeness and skin comfort.34
Trimming
Trimming with scissors is a recommended option to make the hair less noticeable.35
Plucking
Plucking is slow, tedious, painful and hence not appropriate for widespread hypertrichosis in infants or children.35
Waxing
Waxing is an efficient way of plucking vellus hair in all areas of the body. The major disadvantages are discomfort, skin irritation or folliculitis. The regrowth period is two–six weeks.35
Bleaching
Bleaching with 6% hydrogen peroxide oxidizes and softens the hair making it less apparent. However, it can occasionally result in irritation; yellow bleached hair is also highly visible in dark-skinned individuals.35
Chemical depilatories
Chemical depilatory creams comprise salts of thioglycolic and thiolactic acid as the main active ingredient. Thioglycolates are used in a concentration of 2–4% and act within 5–15 min.
Since thioglycolates attack keratin, and the hair shaft and epidermis are similar in their keratin composition, most chemical depilatories hold a high irritancy potential. Their application is messy; they have an unpleasant odor and are relatively expensive, especially if treating larger areas. In children with extensive hypertrichosis, treatment with chemical depilatories should be limited to localized sites because of a theoretical risk of additional toxicity from systemic thioglycolate absorption.35
Electrosurgical epilation
Epilation using the blend method is the most useful method. However, the main disadvantages are the length, number of treatments, risk of scarring, perifollicular inflammation and post-inflammatory hypo- or hyper-pigmentation, making it unsuitable for use in children.35
Topical eflornithine
Eflornithine irreversibly inhibits ornithine decarboxylase, an enzyme that regulates the conversion of ornithine to polyamines (putrescine, spermine and spermidine). Polyamines are thought to play important roles in nucleic acid synthesis, cell division and differentiation. Eflornithine reduces the length of the anagen phase, slowing excessive hair growth. A thin layer of eflornithine hydrochloride 13.9% cream is rubbed in thoroughly over the affected areas twice daily, eight hours apart. Improvement is seen in four–eight weeks, though it may be longer in some patients. It is recommended that eflornithine be stopped after six months if there is no visible improvement. In those who respond, stopping eflornithine, will lead to hair regrowth in eight weeks. Vellus hair which does not respond to laser can be treated with eflornithine. Laser treatment given with eflornithine is more efficacious than eflornithine alone. The safety and efficacy of topical eflornithine for widespread hypertrichosis and in children under 12 years have not been established.36
Lasers and light sources
As these disorders are rare, large studies regarding the safety and tolerability of laser hair removal in children are lacking.
The diode in motion (800–810nm), long-pulsed Q-switch Nd: Yag (1064 nm) with or without topical carbon solution, and intense pulsed light (590–1200 nm) are all safe and well-tolerated in children under 16 years with Fitzpatrick type IIIV when administered properly.37 Young children may require topical anesthesia, but the surface area that can be anesthetized at any one time is limited by the safe maximum dose, which necessitates multiple treatment sessions for large areas such as the back in them. General anesthesia is administered when immobility is required for safety purposes or with extensive treatment areas.38
Authors’ Recommendation
Management includes family screening, genetic counseling, advice regarding the chance of spontaneous remission, and discussion of the temporary methods of hair removal.
For generalized hypertrichosisa.-
Children under 12 years — Trimming or repeated shaving using modern electric shavers followed by application of barrier repair cream
Children 12 years and above — Repeated shaving or trimming or diode in motion (800–810 nm).
For localized hypertrichosis–
Children under 12 years — Repeated shaving or trimming
Children 12 years and above — Repeated shaving or trimming or chemical depilatory or diode in motion (800–810nm) or eflornithine hydrochloride cream (as an adjuvant for facial hyperhidrosis).
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Hair disorders In: Solomon LM, Esterly NP, Loeffel ED, eds. Major Problems in Clinical Pediatrics Adolescent Dermatology. Vol 19. Philadelphia, PA: WB Saunders; 1978. p. :361-6.
- [Google Scholar]
- Hypertrichosis: A report of three cases. Br Med J. 1950;1:1171-4.
- [CrossRef] [PubMed] [Google Scholar]
- Childhood hypertrichosis: Diagnosis and management. Arch Dis Child. 1995;72:457-9.
- [CrossRef] [PubMed] [Google Scholar]
- Congenital hypertrichosis lanuginosa in a father and son. Arch Dermatol. 2011;147:746-7.
- [CrossRef] [PubMed] [Google Scholar]
- A new form of hypertrichosis inherited as an X-linked dominant trait. Hum Genet. 1984;66:66-70.
- [CrossRef] [PubMed] [Google Scholar]
- Prenatally diagnosed partial trisomy 3q case with an omphalocele and less severe phenotype. J Turk Ger Gynecol Assoc. 2010;11:228-32.
- [CrossRef] [PubMed] [Google Scholar]
- Barber-say syndrome and ablepharon-macrostomia syndrome: An overview. Am J Med Genet A. 2016;170:1989-2001.
- [CrossRef] [PubMed] [Google Scholar]
- Cantú syndrome is caused by mutations in ABCC9. Am J Hum Genet. 2012;90:1094-101.
- [CrossRef] [PubMed] [Google Scholar]
- Craniofacial dysostosis, hypertrichosis, genital hypoplasia, ocular, dental, and digital defects: Confirmation of the Gorlin-Chaudhry-Moss syndrome. Am J Med Genet. 1992;44:518-22.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosis and management of Cornelia de Lange syndrome: First international consensus statement. Nat Rev Genet. 2018;19:649-66.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosis of Ambras syndrome: Comments on complex cytogenetic rearrangement of chromosome 8q in a case of Ambras syndrome. Am J Med Genet. 2002;109:77-8.
- [CrossRef] [PubMed] [Google Scholar]
- Lymphedema-Distichiasis syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, eds. GeneReviews®. Seattle, WA: University of Washington; 1993-2020.
- [Google Scholar]
- Rubinstein-Taybi syndrome: Clinical and molecular overview. Expert Rev Mol Med. 2007;9:1-6.
- [CrossRef] [PubMed] [Google Scholar]
- Schinzel-Giedion syndrome: A novel case, review and revised diagnostic criteria. J Genet. 2018;97:35-46.
- [CrossRef] [PubMed] [Google Scholar]
- The stiff skin syndrome case series, differential diagnosis of the stiff skin phenotype, and review of the literature. Arch Dermatol. 2008;144:1351-9.
- [CrossRef] [PubMed] [Google Scholar]
- The progression of Wiedemann-Steiner syndrome in adulthood and two novel variants in the KMT2A gene. Am J Med Genet A. 2019;179:300-5.
- [CrossRef] [PubMed] [Google Scholar]
- Mutations in KCNH1 and ATP6V1B2 cause Zimmermann-Laband syndrome. Nat Genet. 2015;47:661-7.
- [CrossRef] [PubMed] [Google Scholar]
- Decreased half-life of insulin-like growth factor I in Rabson-Mendenhall syndrome. J Inherit Metab Dis. 2001;24:546-50.
- [CrossRef] [PubMed] [Google Scholar]
- Genotype-phenotype correlation in inherited severe insulin resistance. Hum Mol Genet. 2002;11:1465-75.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical course of genetic diseases of the insulin receptor (Type A and Rabson-Mendenhall syndromes): A 30-year prospective. Medicine (Baltimore). 2004;83:209-22.
- [CrossRef] [PubMed] [Google Scholar]
- Leprechaunism-a case report. Indian J Dermatol. 2012;57:499-501.
- [CrossRef] [PubMed] [Google Scholar]
- Lipodystrophy syndromes: Presentation and treatment. In: Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dungan K, eds. Endotext. South Dartmouth, MA: MDText Inc; 2000.
- [Google Scholar]
- Porphyrias Consortium of the NIH-Sponsored Rare Diseases Clinical Research Network Congenital erythropoietic porphyria. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Mirzaa G, eds. GeneReviews®. Seattle, WA: University of Washington; 1993-2020.
- [Google Scholar]
- The H syndrome: Molecular diagnosis using next generation sequencing. AACE Clin Case Rep. 2016;2:e65-9.
- [CrossRef] [Google Scholar]
- The H syndrome: The first 79 patients. J Am Acad Dermatol. 2014;70:80-8.
- [CrossRef] [PubMed] [Google Scholar]
- Leigh and Leigh-like syndrome in children and adults. Pediatr Neurol. 2008;39:223-35.
- [CrossRef] [PubMed] [Google Scholar]
- Neuropathy target esterase impairments cause Oliver-McFarlane and Laurence-Moon syndromes. J Med Genet. 2015;52:85-94.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical hints to diagnosis of attenuated forms of mucopolysaccharidoses. Ital J Pediatr. 2018;44:132.
- [CrossRef] [PubMed] [Google Scholar]
- Diagnosis of mucopolysaccharidosis based on history and clinical features: Evidence from the Bajio REGION of Mexico. Cureus. 2018;10:e3617.
- [CrossRef] [PubMed] [Google Scholar]
- Winchester syndrome: A case report. Int J Dermatol. 2009;48:175-7.
- [CrossRef] [PubMed] [Google Scholar]
- Innovative approaches to avoid electric shaving-induced skin irritation. Int J Cosmet Sci. 2016;38(Suppl 1):10-6.
- [CrossRef] [PubMed] [Google Scholar]
- Causes and management of hypertrichosis. Am J Clin Dermatol. 2002;3:617-27.
- [CrossRef] [PubMed] [Google Scholar]
- Eflornithine. Indian J Dermatol Venereol Leprol. 2007;73:365-6.
- [CrossRef] [PubMed] [Google Scholar]
- Laser hair removal in a patient with hypertrichosis lanuginosa congenita. Dermatol Surg. 1997;23:705-7.
- [CrossRef] [PubMed] [Google Scholar]
- Hair removal with long-pulse alexandrite and long-pulse Nd: YAG lasers is safe and well tolerated in children. Clin Experim Dermatol. 2009;34:684-7.
- [CrossRef] [PubMed] [Google Scholar]




