
Translate this page into:
Acromegaloid facial appearance with generalised hypertrichosis: A novel phenotype of AFF4 mutation

Corresponding author: Dr. Rahul Mahajan, Department of Dermatology, Postgraduate Institute of Medical Education and Research, Chandigarh, India. drrahulpgi@yahoo.com
-
Received: ,
Accepted: ,
How to cite this article: Singh S, Mehta H, Kumar A, Dogra S, Mahajan R. Acromegaloid facial appearance with generalised hypertrichosis: A novel phenotype of AFF4 mutation. Indian J Dermatol Venereol Leprol. doi: 10.25259/IJDVL_624_2023
Dear Editor,
Acromegaloid facial appearance (AFA) syndrome is a rare disorder characterised by coarse facies with a bulbous nose, thickened lips, and overgrowth of the oral mucosa. It has been described in association with hypertrichosis in the previous reports with no genetic loci identified so far. 1 Herein we describe a case presenting with the phenotype of AFA syndrome with previously undescribed distinct mutations in the AFF4 gene.
A 15 yr male child, born to parents unrelated by blood, presented with a history of generalised hypertrichosis since early childhood. Examination of the face revealed prominent metopic and supraorbital ridges with prominent brows, underdeveloped nasolabial folds, a short chin, coarse facies with heavy and rounded features, thickened skin, and prominent bulbous nose, thickened lips, gingival hyperplasia and generalised hypertrichosis [Figures 1 and 2]. Systemic examination was normal. He denied intake of any medications. There was no significant family history. He was born by a normal vaginal delivery without any complications. He was detected to have glucose-6-phosphate dehydrogenase (G6-PD) deficiency since birth. The child had normal developmental milestones with no intellectual disability. Keeping in mind the clinical picture, differentials of AFA syndrome, congenital generalised hypertrichosis terminalis, and CHOPS (cognitive impairment and coarse facies, heart defects, obesity, pulmonary involvement, short stature, and skeletal dysplasia) syndrome were considered and further evaluation was performed.

- Prominent supraorbital ridges with prominent brows, underdeveloped nasolabial folds, short chin, coarse face with heavy and rounded features, thickened skin and prominent bulbous nose, thickened lips, gingival hyperplasia, and generalised hypertrichosis.
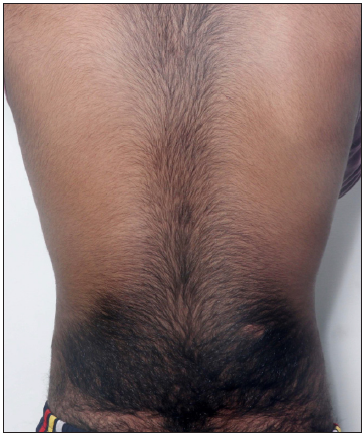
- Generalised hypertrichosis showing coarse terminal hair over the back.
Contrast-enhanced computed tomography (CECT) of the chest to detect pulmonary involvement and a bone scan to evaluate skeletal deformities revealed no abnormalities. Echocardiography of the heart showed a small patent foramen ovale (PFO) without any functional limitation. The endocrine evaluation was within the normal limits. The skeletal radiographs showed no abnormality. Histopathological examination of biopsies obtained from lips, gums, and nose revealed similar findings of pseudoepitheliomatous hyperplasia with minimal perivascular infiltrate without any granulomas or dysplasia. Whole exome sequencing revealed a missense mutation (a C to A substitution at nucleotide (nt) 130) that results in a heterogeneous substitution of Valine for Glycine at codon 130 (Gly130Val) in exon 3 of the AFF4 gene. The patient had a phenotype that was consistent with AFA syndrome; however, the genetic mutation in the AFF4 gene was inconsistent with the same.
AFA syndrome is a rare, complex genetic condition of unknown origin. It is characterised by distinct acromegaloid facial features, including thickened lips, overgrowth of oral mucosa with pronounced rugae, a bulbous nose, and pronounced upper eyelids, resulting in constriction of the palpebral fissure. 1 After excluding excess of growth hormone and insulin resistance, it is important to consider the possibility of AFA syndrome in the list of potential diagnoses. In addition, there have been reported cases of AFA syndrome occurring in conjunction with pachydermoperiostosis, cutis verticis gyrata, and keratitis. 2 While no definite genetic locus or chromosomal abnormality has been identified in this syndrome, a few reports suggest autosomal dominant inheritance. In one family affected by AFA syndrome, authors observed a low positive LOD (logarithm of the odds) score for potential linkage between the disorder and chromosome regions 1p, 6p, 14q, and 16q. 3
Congenital generalised hypertrichosis terminalis, accompanied by gingival hyperplasia, is a rare autosomal dominant disorder associated with a microdeletion on chromosome 17q. The disorder is distinguished by the development of terminal hair throughout the entire body, except for the palms, soles, and mucosal areas. In addition, some patients may exhibit constricting bands on their upper limbs as a characteristic feature. 4
CHOPS syndrome is a rare disorder described in the literature with only a few case reports. This condition presents with a range of craniofacial dysmorphic features, including but not limited to synophrys (fusion of eyebrows), upturned nose, arched eyebrows, long eyelashes, facial fullness, and coarse facies. 5 Affected individuals also tend to have short stature and may be obese. Cardiac involvement has also been described in 72.7% of patients with patent ductus arteriosus (PDA) being the most common, followed by a ventricular septal defect (VSD) and PFO. 5 Pulmonary involvement is also commonly described in the form of chronic lung disease, subglottic stenosis, pulmonary haemorrhage, and sleep apnoea. The spectrum of skeletal abnormalities described with CHOPS syndrome includes vertebral anomalies such as decreased height or fusion of vertebral bodies, hip subluxation, and brachydactyly. Developmental delay and neurocognitive decline were other observed features. The mutations in CHOPS syndrome are spontaneous and occur in 14 amino-acid conserved regions of the AFF4 protein, resulting in impaired proteasomal degradation which contributes to the rarity of the disease. 6
While AFA syndrome is well documented in the literature and typically characterised by coarse facial features, mucosal thickening and hypertrichosis, our case presents unique features. It includes dysmorphic traits and a novel missense mutation in the AFF4 gene which, to our current knowledge, has not been reported in AFA syndrome before. Further functional studies are needed to establish a potential correlation between AFF4 pathway and epidermal hyperplasia and hypertrichosis.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Acromegaloid facial appearance syndrome - A new case in India. J Genet Med. 2013;10:57-61.
- [Google Scholar]
- Acromegaloid facial appearance and hypertrichosis: A case suggesting autosomal recessive inheritance. Clin Dysmorphol. 2004;13:49-50.
- [CrossRef] [PubMed] [Google Scholar]
- An autosomal dominant syndrome of acromegaloid facial appearance and generalised hypertrichosis terminalis. J Med Genet. 1996;33:972-74.
- [CrossRef] [PubMed] [Google Scholar]
- Congenital generalized hypertrichosis, gingival hyperplasia, a coarse facies with constriction bands: A rare association. Int J Trichology. 2015;7:67-71.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Clinical and molecular spectrum of CHOPS syndrome. Am J Med Genet. 2019;179:1126-38.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Germline gain-of-function mutations in AFF4 cause a developmental syndrome functionally linking the super elongation complex and cohesin. Nat Genet. 2015;47:338-44.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




