
Translate this page into:
Bathing-suit ichthyosis: A rare but valuable entity for developing personalised pathogenesis-based therapies for autosomal recessive congenital ichthyosis
*contributed equally
Corresponding author: Dr. Vamsi Krishna Yenamandra, Division of Disease Biology, CSIR-Institute of Genomics & Integrative Biology, New Delhi, India; Academy of Scientific and Innovative Research, Ghaziabad, India. vamsi.krishna@igib.res.in
-
Received: ,
Accepted: ,
How to cite this article: Tandon S, Konda SC, Kola R, Sowpati DT, Tallapaka KB, Yenamandra VK. Bathing-suit ichthyosis: A rare but valuable entity for developing personalised pathogenesis-based therapies for autosomal recessive congenital ichthyosis. Indian J Dermatol Venereol Leprol. doi: 10.25259/IJDVL_954_2024
Dear Editor,
‘Bathing-suit’ ichthyosis (BSI) is a rare and mild form of autosomal recessive congenital ichthyosis (ARCI), clinically characterised by a restricted distribution of dark adherent skin scales.1 In-situ and in-vitro studies, complemented with digital thermal imaging, deduced that BSI is caused by pathogenic variants in the TGM1 (transglutaminase-1) gene that decrease the thermostability of the encoded enzyme, particularly at warmer regions of the body.1,2 However, such phenotype-genotype correlations are not always straightforward, as the same pathogenic variants can cause other severe ARCI forms, including a dynamic shift from the BSI phenotype over time and vice-versa.3 Nevertheless, in the context of incurable diseases like ARCI, identifying naturally occurring mild phenotypes and delving deeper into their underlying molecular mechanisms might provide innovative leads for the development of affordable and indigenous therapeutics. In countries like India, where research studies are largely limited to academic institutions, developing such patient cohorts is challenging due to limited awareness, expertise, financial constraints, and patients with mild phenotypes rarely availing of medical services. Towards this, we wish to report a genetically characterised BSI patient identified at a dermatology outpatient clinic through our collaborative genomics program and provide additional insights that could improve our current understanding of BSI pathogenesis.
A 14-year-old boy, born of consanguineous marriage, was referred to us for genetic evaluation with a provisional diagnosis of mild ARCI and a history of collodion membrane at birth. After obtaining informed consent, whole exome sequencing was performed.4 Variant analysis revealed a homozygous ‘likely-pathogenic’ missense variant in the TGM1 gene (NM_000359.3: exon3:c.391A>G; p.Thr131Ala). This variant was predicted to be deleterious by in-silico softwares, absent in publicly available population databases, including the Indian population, and was previously reported only once in an Asian patient with an unclassified ARCI phenotype.5 No other ‘pathogenic’ variants were identified in any other known ARCI genes. Clinical re-evaluation of our patient revealed the presence of dark adherent scales restricted to the trunk and neck, with distinct sparing of the face and extremities, confirming a characteristic BSI phenotype [Figure 1]. Further, sanger validation along with family segregation analysis confirmed an autosomal recessive inheritance, and the patient was referred for genetic counselling and enrolled in a follow-up study to understand molecular consequences of the identified variant.
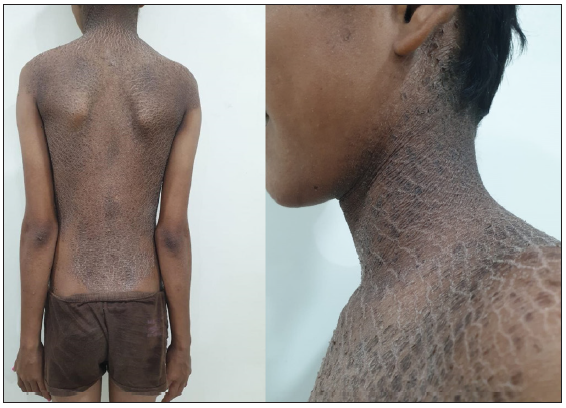
- Dark adherent scales on the trunk and neck along with sparing of face and extremities.
Literature analysis revealed that only 72 genetically characterised BSI patients were reported worldwide, a majority of whom had unique combinations of compound heterozygous TGM1 pathogenic variants, particularly involving ‘arginine’ substitutions in at least one allele (85%, n = 61/72), notably at Arg307 and Arg315 (63%, n = 45/72) residues in the catalytic core domain [Figure 2]. Contrary to this general trend, our patient showed a homozygous ‘threonine’ substitution (Thr131Ala) within the beta-sandwich domain (crucial for protein folding and enzyme stability), adding to the growing list of non-arginine substitutions responsible for the BSI phenotype. Structurally, Thr131 is a highly conserved hydrophilic, polar residue that forms protein-stabilising contacts at the domain interface, like hydrogen bonds (with Arg126, Tyr134, and Ile140) and Van der Waals interactions (with Arg126, His129, His130, Tyr134, Ile140. and Arg142), involving surrounding residues. Substitution of Thr131 with ‘alanine’ (a hydrophobic, non-polar residue with a compact side chain) disrupts these stabilising contacts, particularly with Arg142, resulting in an altered surface loop that is susceptible to abnormal protein folding and increased proteolytic cleavage [Figure 3]. Although the lack of TGM1 enzyme studies preclude us from ascertaining functional significance to the identified variant, such preliminary findings about structural consequences coupled with previous literature evidence shows that severe ARCI phenotypes are more likely to occur due to alterations at these interacting residues [Figure 2].6 This raises the question whether the BSI phenotype noted in our patient was an unlikely event.
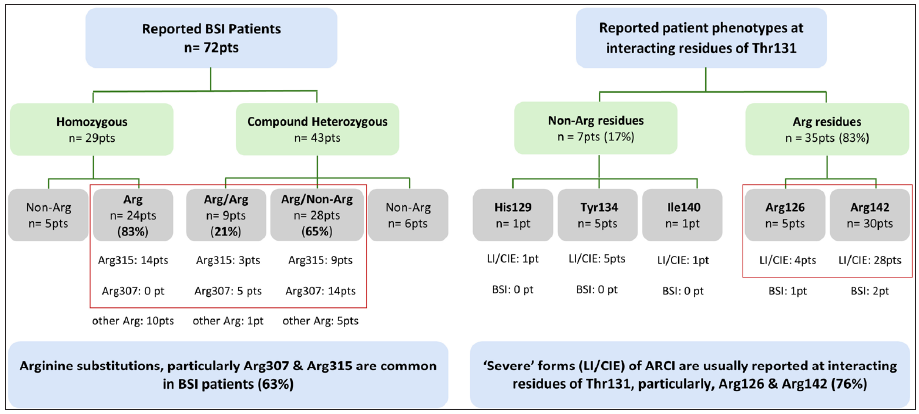
- Trends in genotypic alterations in previously reported BSI patients and reported phenotypes associated with the interacting partners of Thr131 (mutated residue in our patient), primarily involving severe forms like lamellar ichthyosis (LI) and congenital ichthyosiform erythroderma (CIE).
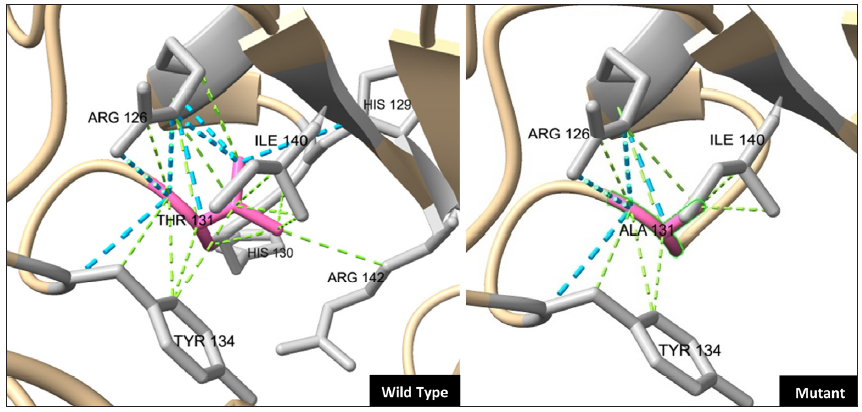
- The computational wild-type TGM1 protein model from Google AlphaFold (ID: P22735), was utilised as a template to generate a mutant protein model (with the p.Thr131Ala variant) and to study interacting partners using the in-built mutagenesis and structure analysis features of the UCSF ChimeraX tool (v1.6.1, https://www.rbvi.ucsf.edu/chimerax). The crucial interacting residues (in grey) are shown as ball and stick and the stabilising contacts (Hydrogen and Van der Waals bonds) are shown as blue and green dotted lines, respectively.
Given that a majority of BSI-causing TGM1 variants involve arginine residues at CpG sites and/or NCG-GNN consensus motif,3,6 it is plausible that epigenetic regulatory mechanisms like methylation/demethylation (reported to regulate TGM1 expression7) processes might indeed be playing a dominant role in BSI pathogenesis, in addition to reduced thermostability. Bisulfite sequencing using patient skin biopsies from affected and unaffected regions might help in discovering unique molecular signatures, if any. While these studies are planned in our patient, herein we wish to highlight the importance of identifying such naturally occurring mild phenotypes through collaborative genomics programs. Rapid advances in genomic technologies are enabling us to address complex questions beyond the regular phenotype-genotype correlations, and making them accessible might benefit the development of indigenous pathogenesis-based therapies for incurable diseases like ARCI.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
ST acknowledges fellowship support from Council of Scientific and Industrial Research (CSIR) through award no. GATE31/0043(13267)/2022-EMR-I; VKY acknowledges funding support from Council of Scientific and Industrial Research (CSIR), through grant no. OLP1164.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Bathing suit ichthyosis is caused by transglutaminase-1 deficiency: Evidence for a temperature-sensitive phenotype. Hum Mol Genet. 2006;15:3083-97.
- [CrossRef] [PubMed] [Google Scholar]
- Transglutaminase-1 and bathing suit ichthyosis: Molecular analysis of gene/environment interactions. J Invest Dermatol. 2009;129:2068-71.
- [CrossRef] [PubMed] [Google Scholar]
- Bathing suit ichthyosis: Two burmese siblings and a review of the literature. Pediatr Dermatol. 2020;37:165-70.
- [CrossRef] [PubMed] [Google Scholar]
- Genotype-phenotype correlations of dystrophic epidermolysis bullosa in India: Experience from a tertiary care centre. Acta Derm Venereol. 2018;98:873-9.
- [CrossRef] [PubMed] [Google Scholar]
- The Genomic and phenotypic landscape of ichthyosis: An analysis of 1000 kindreds. JAMA Dermatol. 2022;158:16-25.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Transglutaminase-1 gene mutations in autosomal recessive congenital ichthyosis: Summary of mutations (including 23 novel) and modeling of TGase-1. Hum Mutat. 2009;30:537-47.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- GRHL3/GET1 and trithorax group members collaborate to activate the epidermal progenitor differentiation program. PLoS Genet. 2012;8:e1002829.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




