
Translate this page into:
An unusual cutaneous presentation of neurofibromatosis type 2 with clinically silent vestibular schwannomas following a pathogenic nonsense mutation in NF2 gene
Corresponding author: Dr. Betsy Ambooken, Department of Dermatology and Venereology, Government Medical College, Thrissur, Mulangunnathukavu, Thrissur, India. joebetsy123@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Ambooken B, Rajeev R, Asokan N, Ananda Kesavan TM. An unusual cutaneous presentation of neurofibromatosis type 2 with clinically silent vestibular schwannomas following a pathogenic nonsense mutation in NF2 gene. Indian J Dermatol Venereol Leprol. doi: 10.25259/IJDVL_934_2024
Dear Editor,
Neurofibromatosis type 2 (NF2) is an autosomal dominant disease with less profuse cutaneous manifestations when compared to NF1.1 We report a child with florid cutaneous manifestations without overt neurological, ocular or auditory involvement.
A ten-year-old female child presented with multiple painful swellings, gradually increasing in size and number from two years of age. She was born to non-consanguineous parents, and her milestones were normal. Features suggestive of neurocutaneous syndrome were not observed in her family members. On examination, there were multiple skin-coloured, pink, tender, sessile and pedunculated nodules distributed one each on the scalp, lower back, and right shoulder [Figures 1a, 1b] and multiple small sessile lobulated papules on the left forearm. A large linear hypopigmented macule with serrated borders (16 × 4 cm) was seen extending from the umbilicus to the left side of the chest wall with well-preserved margins on diascopy and erythema on rubbing of the skin. A pink pedunculated nodule was seen on the lower border of the hypopigmented macule [Figure 1c]. She also had a linear skin-coloured nodule along the medial border of the left index finger [Figure 1d]. There were three café au lait macules (CALMs) with sizes varying from 1–3 cm on the trunk with feathery margins [Figure 1e]. A detailed systemic examination, including a neurological, ocular and otorhinolaryngological evaluation was normal.
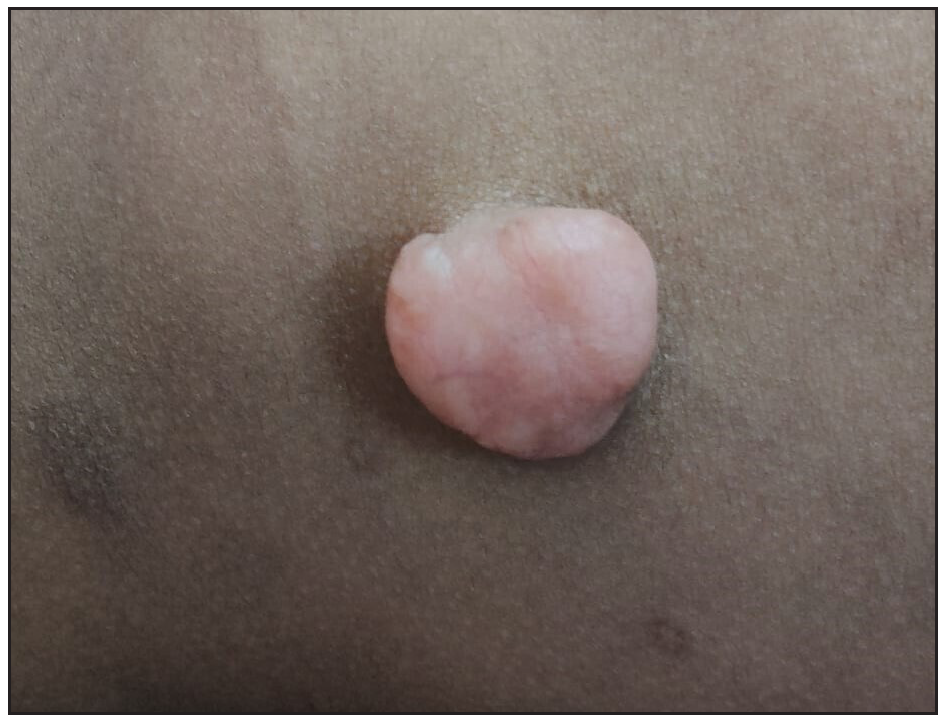
- Pedunculated pink nodule with telangiectasia on the lower back.
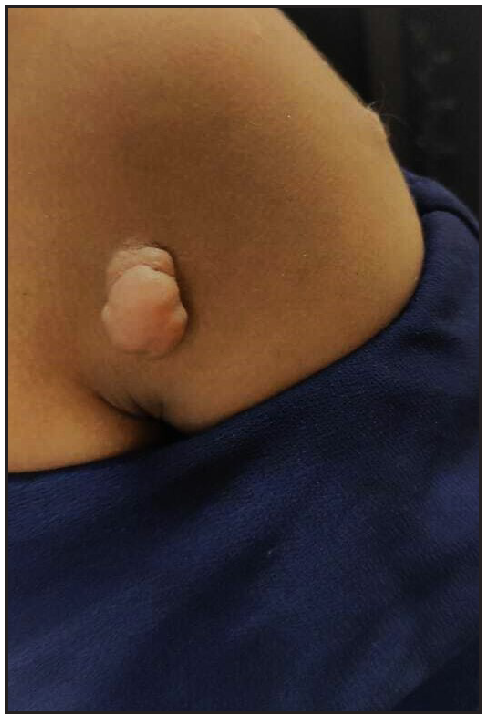
- Pedunculated skin-coloured nodule on the right shoulder.
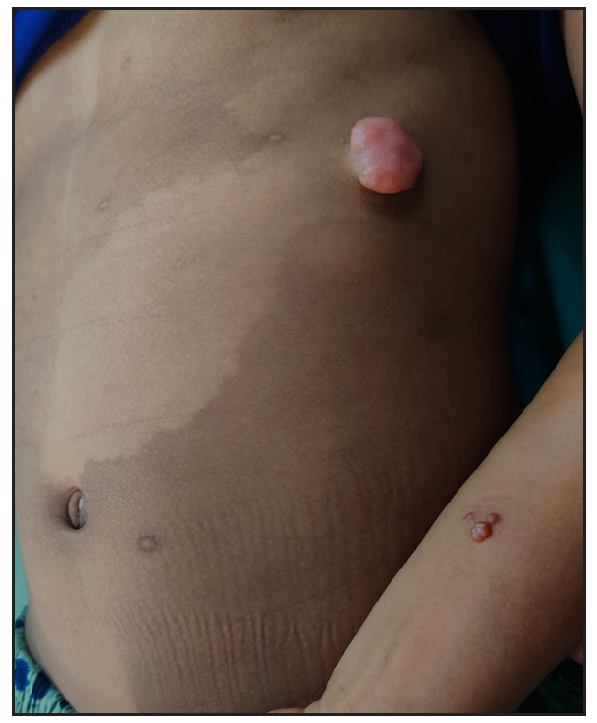
- A large naevus depigmentosus on the left side of the chest wall with a pedunculated nodule on its lower border and sessile nodules on the left forearm.
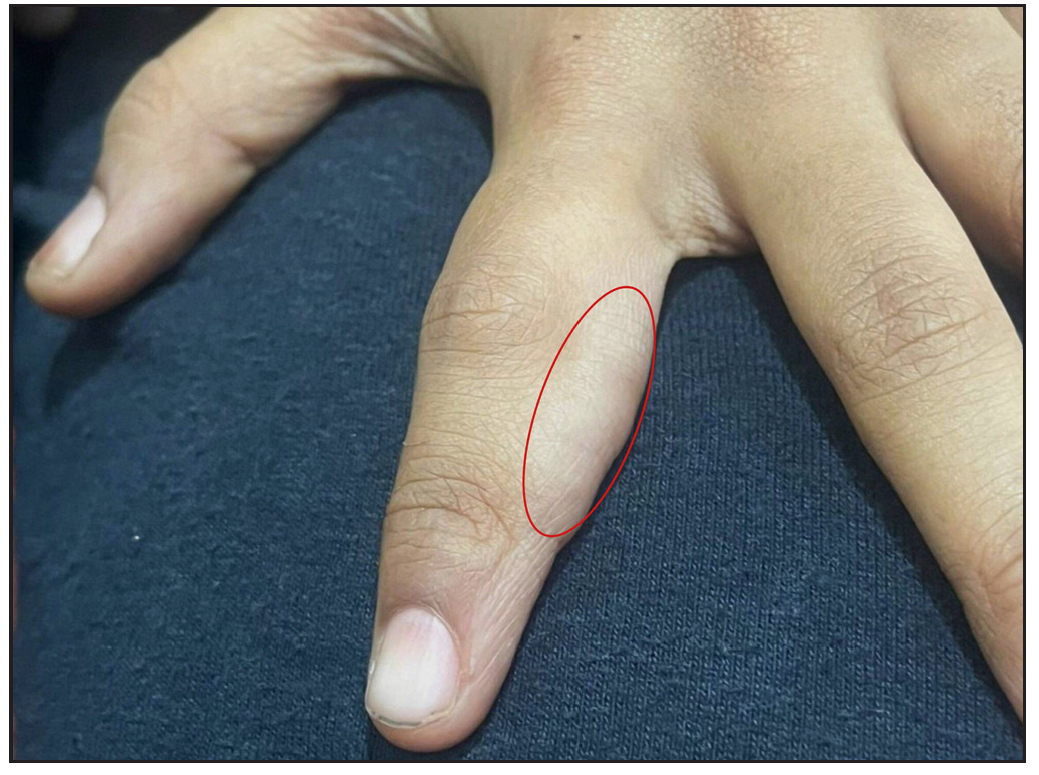
- Linear skin-coloured nodule on the medial border of the left index finger seen within the red circle.
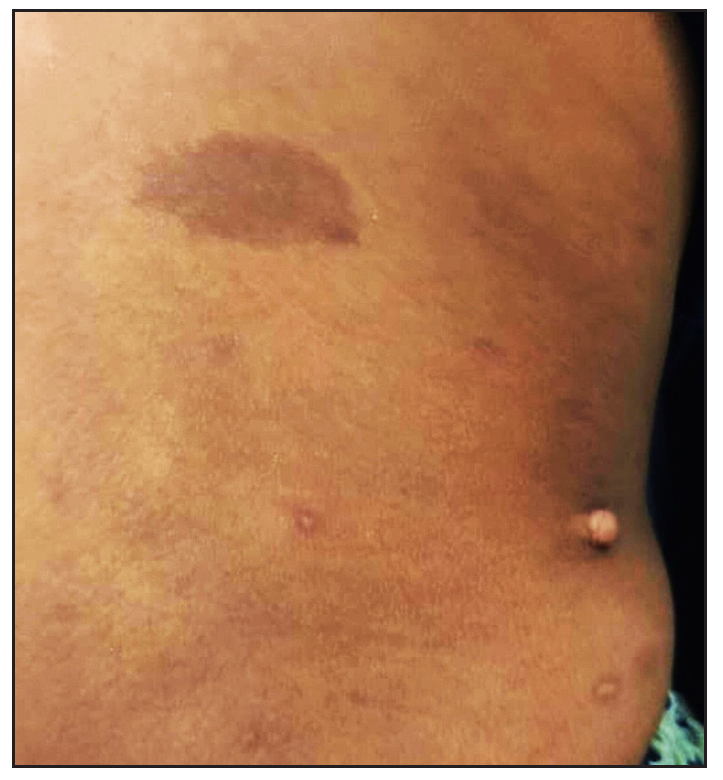
- Café au lait macule with feathery borders and a sessile schwannoma on the back of the trunk.
Skin biopsy revealed an encapsulated nodule in the reticular dermis composed of spindle-shaped cells with elongated wavy nuclei. Dense palisading of spindle-shaped nuclei with a central pink anucleated zone suggestive of Antoni A body was diagnostic of schwannoma [Figures 2a, 2b]. A brain MRI with gadolinium enhancement showed bilateral vestibular schwannomas (VS) and a solitary meningioma [Figure 3]. Genetic testing by the next-generation sequencing (NGS) of the clinical exome panel revealed a heterozygous nonsense variant in exon 8 of the NF2 gene (chr22:g.29661313C>T at a depth of 169x) that resulted in a stop codon and premature truncation of the protein at codon 262. This variant was reported previously and classified as pathogenic (PVS1, PM2, PP5) in the Clinvar database.
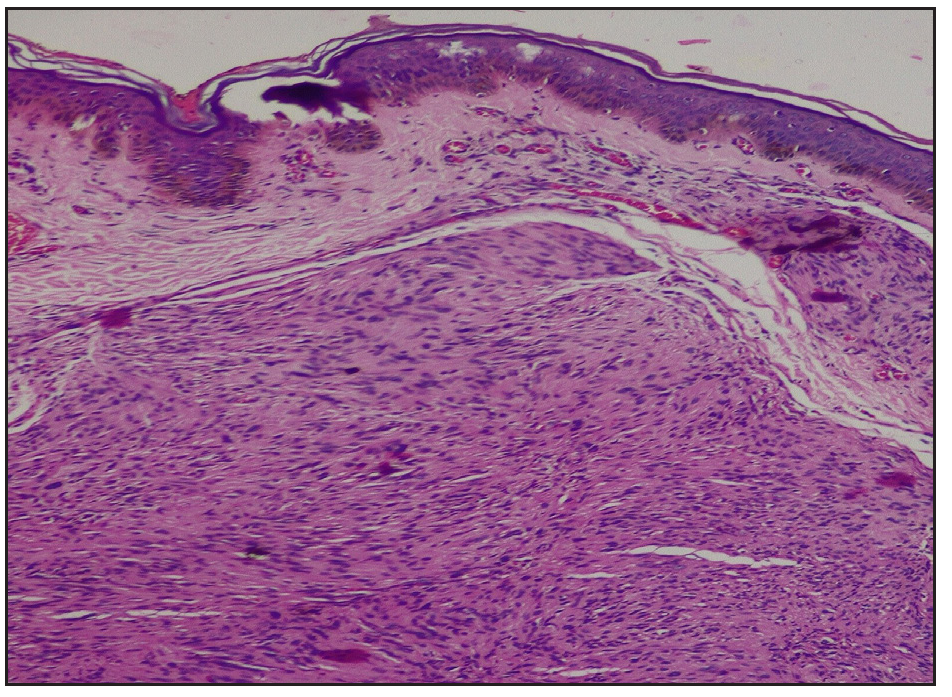
- Reticular dermis showing a neoplasm composed of spindle-shaped cells showing central anuclear zone with peripheral nuclear palisading (Haematoxylin and eosin,100x).
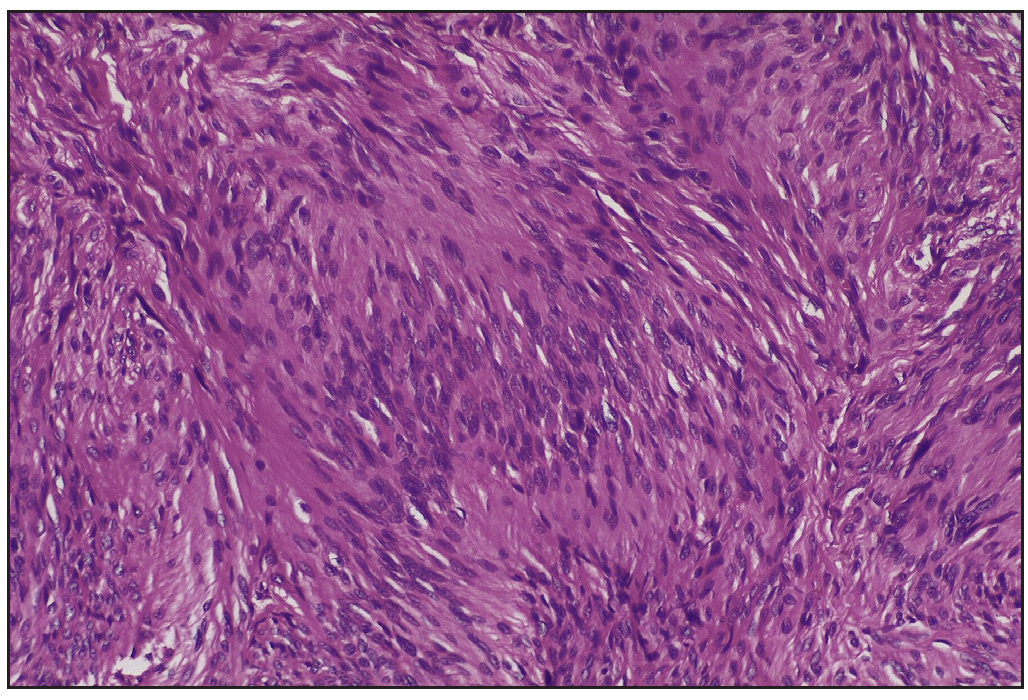
- Verocay bodies with the central anuclear zone (Antoni A) (Haematoxylin and eosin, 400x).
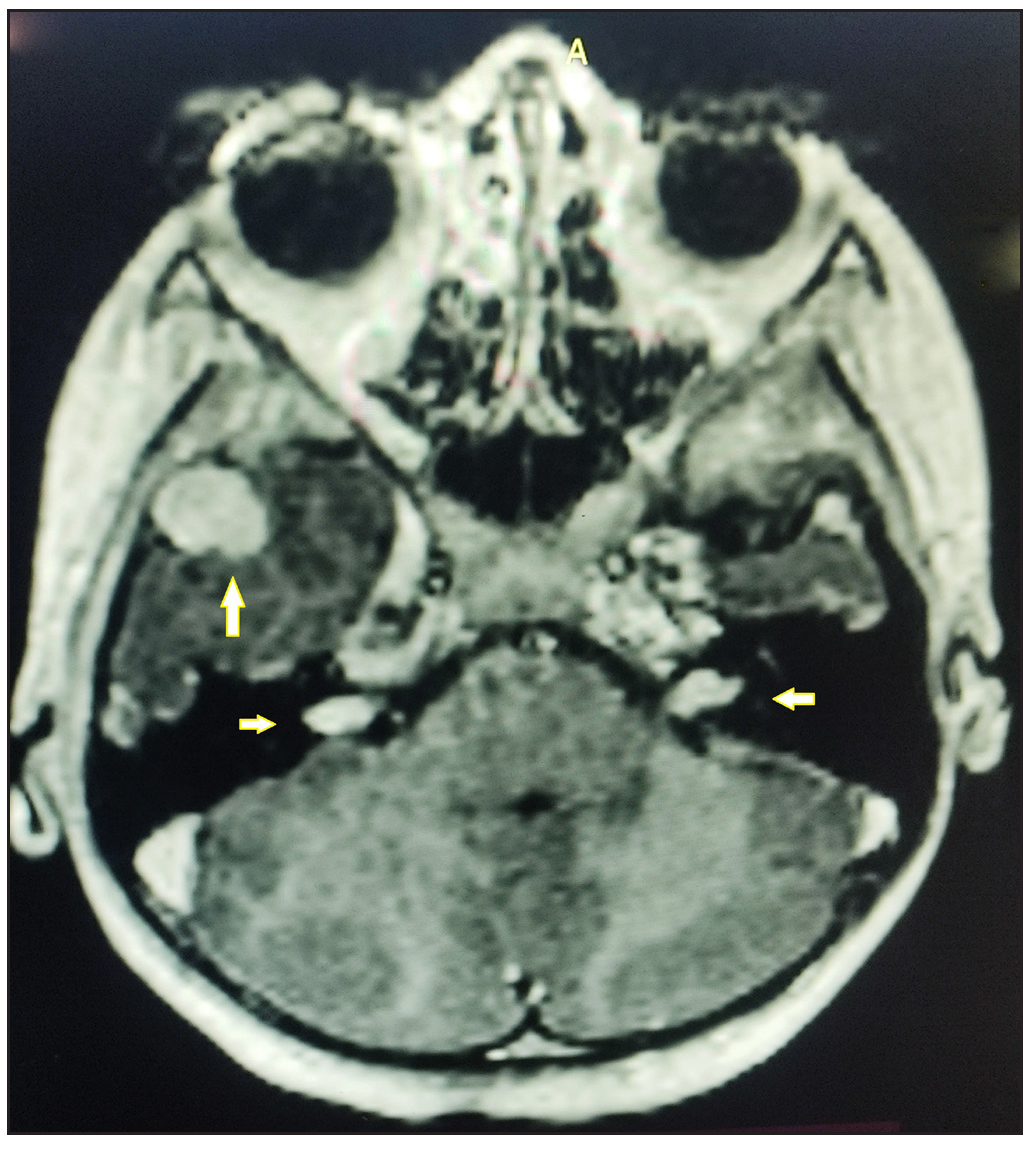
- Brain MRI shows bilateral vestibular schwannomas (horizontal arrows) and meningioma in the right middle cranial fossa (vertical arrow).
Cutaneous schwannomas occur more frequently in NF2 and less often in NF1 and Schwannomatosis (SWN). Hence, all three were considered in the differential diagnosis. Neuroimaging and genetic studies confirmed the diagnosis of NF2.
Historically, NF2 was categorised into two clinical subtypes: congenital Wishart or prepubertal clinical and adult-onset Gardner subtypes.2 This child had an early onset with extensive skin lesions mimicking the Wishart type. Cafe au lait macules (CALMs) in NF2 are less common and fewer in number when compared to NF1.3 Although CALMs were present in this case, they were fewer in number and with indistinct or feathery borders. The presence of cutaneous and subcutaneous schwannomas is a clue to early diagnosis of NF2 and predictive of a more severe phenotype.3 Multiple hypopigmented macules have been previously reported in paediatric cases of NF2.4 The index case had a single large linear Hypomelanotic macule suggestive of naevus depigmentosus with a pedunculated schwannoma, possibly suggestive of a binary genodermatoses.5 However, further genetic studies on tissue samples may be needed to delineate the exact pathogenetic mechanism.
The 1997 Manchester criteria was the most commonly used criteria to diagnose NF2.6 Though there was no family history of NF2, the presence of bilateral VS by itself fulfilled the diagnostic criteria for NF2. Due to overlapping phenotypes, there has been a revision in the diagnostic criteria of NF2 and schwannomatosis (SWN).1 The NF2 gene is identified on chromosome 22q12 that encodes for merlin, a tumour suppressor protein related to ezrin-radixin-moesin that modulates the activity of PI3K/AKT, Raf/MEK/ERK, and mTOR signalling pathways.7 SWN caused by mutations in SMARCB1 or LZTR1 genes are also located on chromosome 22. Hence, genetic testing must be done to differentiate NF2-related Schwannomatosis (SWN) from SMARCB1-related SWN and LZTR1-related SWN.7 Truncating mutations, as observed in this case, are the most frequent germline mutation in NF2, with younger age at onset and causing more severe disease.1
There are no established effective treatments for NF2 patients because tumours are likely to regrow after surgical resection. In this child, cutaneous schwannomas, which were painful, were excised. As the VS were asymptomatic, she is being followed up closely and advised neuroimaging every year. Bevacizumab and everolimus have a therapeutic role for progressive VS and meningiomas in NF2 patients.1
The unique features in this child with clinically silent VS include the early onset of painful sessile, pedunculated, and subcutaneous schwannomas and CALMs with feathery margins along with a paired occurrence of naevus depigmentosus and pedunculated schwannoma.
Acknowledgement
We thank Dr Priyadarshini Pande, Medgenome Lab for help rendered in the interpretation of genetic analysis.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of AI-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- Current understanding of neurofibromatosis type 1, 2, and schwannomatosis. Int J Mol Sci. 2021;22:5850.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Genomics, epigenetics, and hearing loss in neurofibromatosis type 2. Otol Neurotol. 2020;41:e529-37.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Skin lesions in neurofibromatosis type 2: Diagnostic and prognostic significance of cutaneous (plexiform) schwannomas. J Eur Acad Dermatol Venereol. 2022;36:1632-40.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Neurofibromatosis type 2 (NF2): A clinical and molecular review. Orphanet J Rare Dis. 2009;4:16.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Mosaicism in human skin: Understanding Nevi, Nevoid skin disorders and cutaneous neoplasia Berlin. Springer; 2014.
- Neurofibromatosis type 2. Lancet. 2009;373:1974-86.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Updated diagnostic criteria and nomenclature for neurofibromatosis type 2 and schwannomatosis: An international consensus recommendation. Genet Med. 2022;24:1967-77.
- [CrossRef] [PubMed] [Google Scholar]




