Translate this page into:
Two novel mutations of NF1 gene identified in Chinese patients with severe neurofibromatosis type 1
2 Department of Dermatology, Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
3 Department of Dermatology, Fourth Affiliated Hospital, Zhejiang University School of Medicine, Yiwu, Zhejiang, China
4 Department of Plastic Surgery, Second Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China
Correspondence Address:
Yunqing Ren
Department of Dermatology, Second Affiliated Hospital, Zhejiang University School of Medicine, No. 88 Jiefang Road, Hangzhou, Zhejiang 310009
China
| How to cite this article: Zheng Y, Qiu G, Dai H, Zhu C, Xue D, Ren Y. Two novel mutations of NF1 gene identified in Chinese patients with severe neurofibromatosis type 1. Indian J Dermatol Venereol Leprol 2020;86:76-81 |
Sir,
Neurofibromatosis type 1 (NF1; OMIM# 162200) is a common autosomal dominant genetic disorder, with an incidence of 1 in 2000–5000 individuals.[1] Its clinical features include 'café-au-lait' macules (CALMs), multiple neurofibromas, axillary or inguinal freckling, iris Lisch nodules, skeleton anomalies, learning disabilities, and predisposition to malignancies. The mutations responsible for NF1 have been identified in the NF1 gene on chromosome 17q11.2. This gene spans approximately 350 kb of genomic DNA and encodes a large GTPase-activating protein called neurofibromin, which is a negative regulator of the Ras-mediated signal transduction pathway.[2] Until now, the germline mutation rate of the NF1 gene is one of the highest known for human inherited diseases.[3]
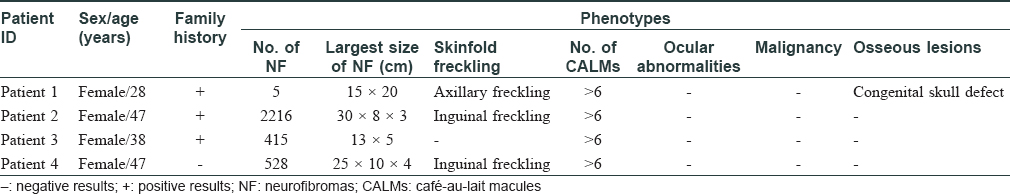
In this study, we performed mutation analysis of the NF1 gene in three Chinese families and in one sporadic case with NF1. The patients were diagnosed based on clinical and histopathological features, according to the National Institutes of Health criteria [Table - 1].[4] All the three probands and the sporadic patient presented with typical skin manifestations, including numerous neurofibromas, especially large size neoplasm, skin freckling, and CALMs all over the body [Figure - 1]. Detailed clinical descriptions of the patients are summarized in [Table - 2].

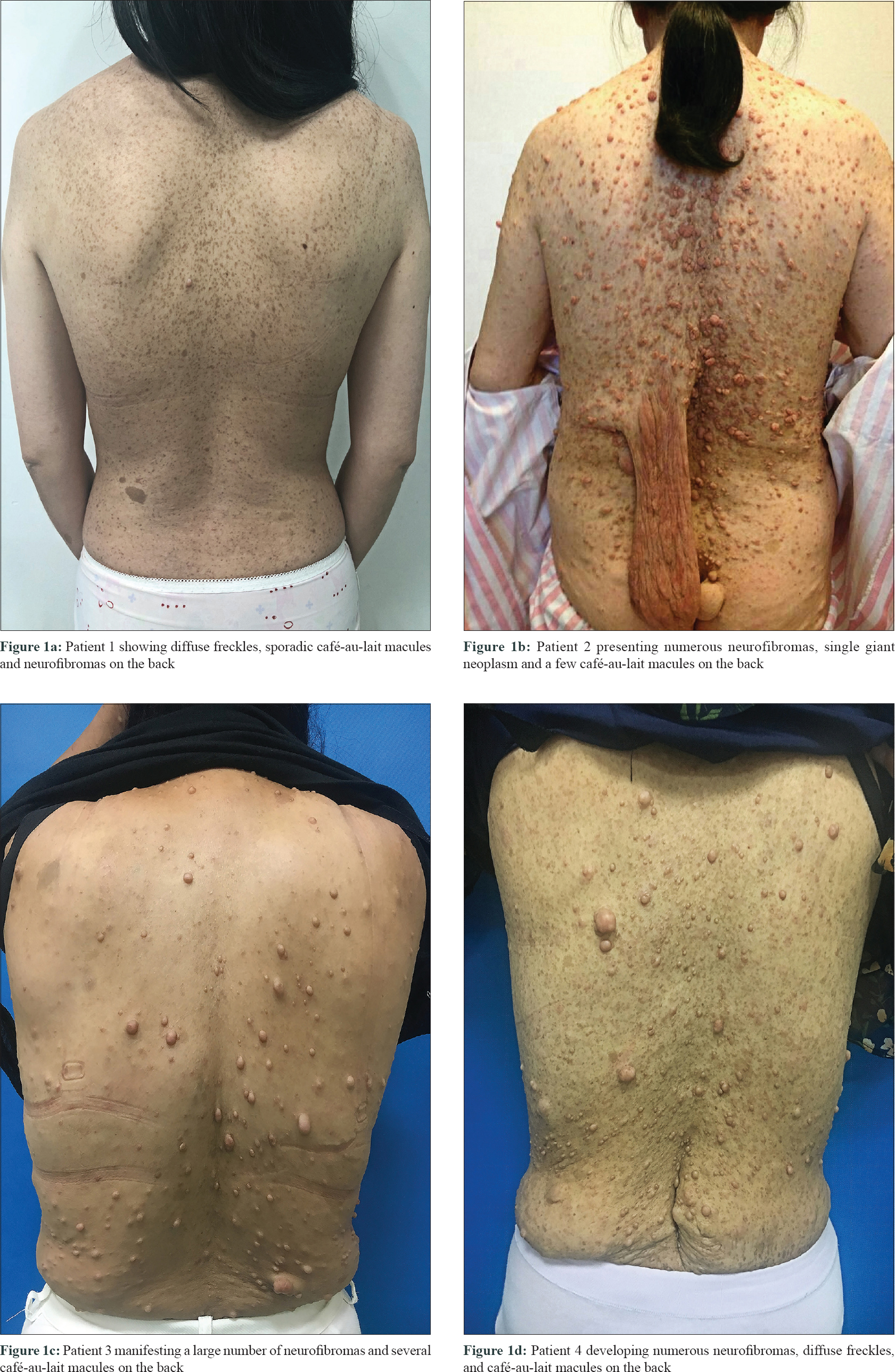 |
| Figure 1: |
The study was approved by the Ethics Committee of Zhejiang University School of Medicine. After proper written informed consent from the participants, blood samples were obtained from the three NF1 families, including 8 affected and 3 unaffected individuals, one sporadic patient and 100 unrelated controls. Genomic DNA was extracted from the peripheral blood using the FlexiGene DNA kit (QIAGEN, Germany). We then sequenced the NF1 gene in four patients using Sanger sequencing, multiplex ligation-dependent probe amplification (MLPA), and complementary DNA (cDNA) sequencing. Four mutations of NF1 gene (NM_000267.3) were identified in our patients, including two novel and two reported mutations [Table - 3] and [Figure - 2].
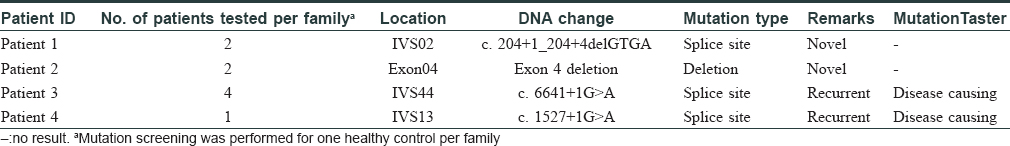
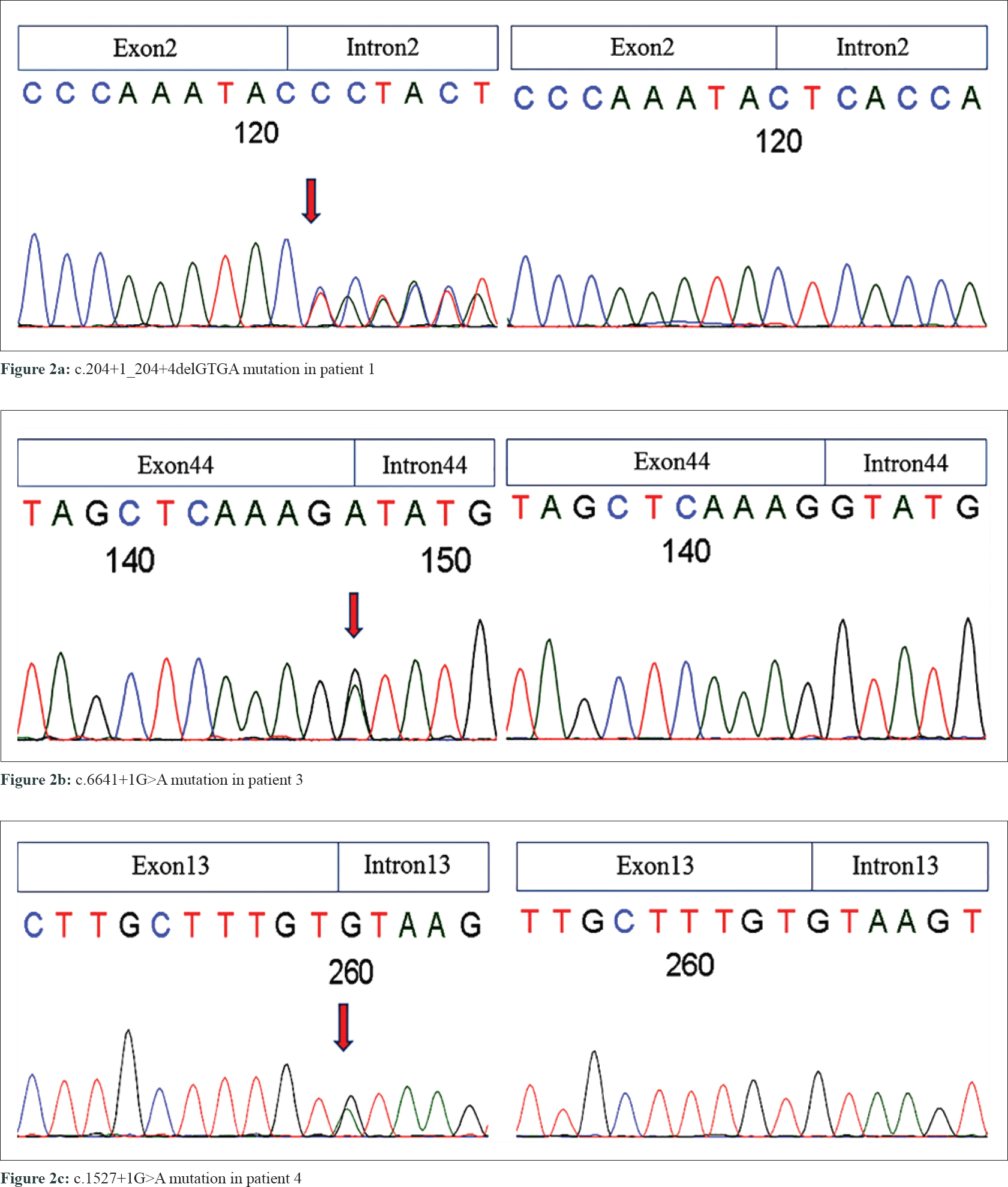 |
| Figure 2: |
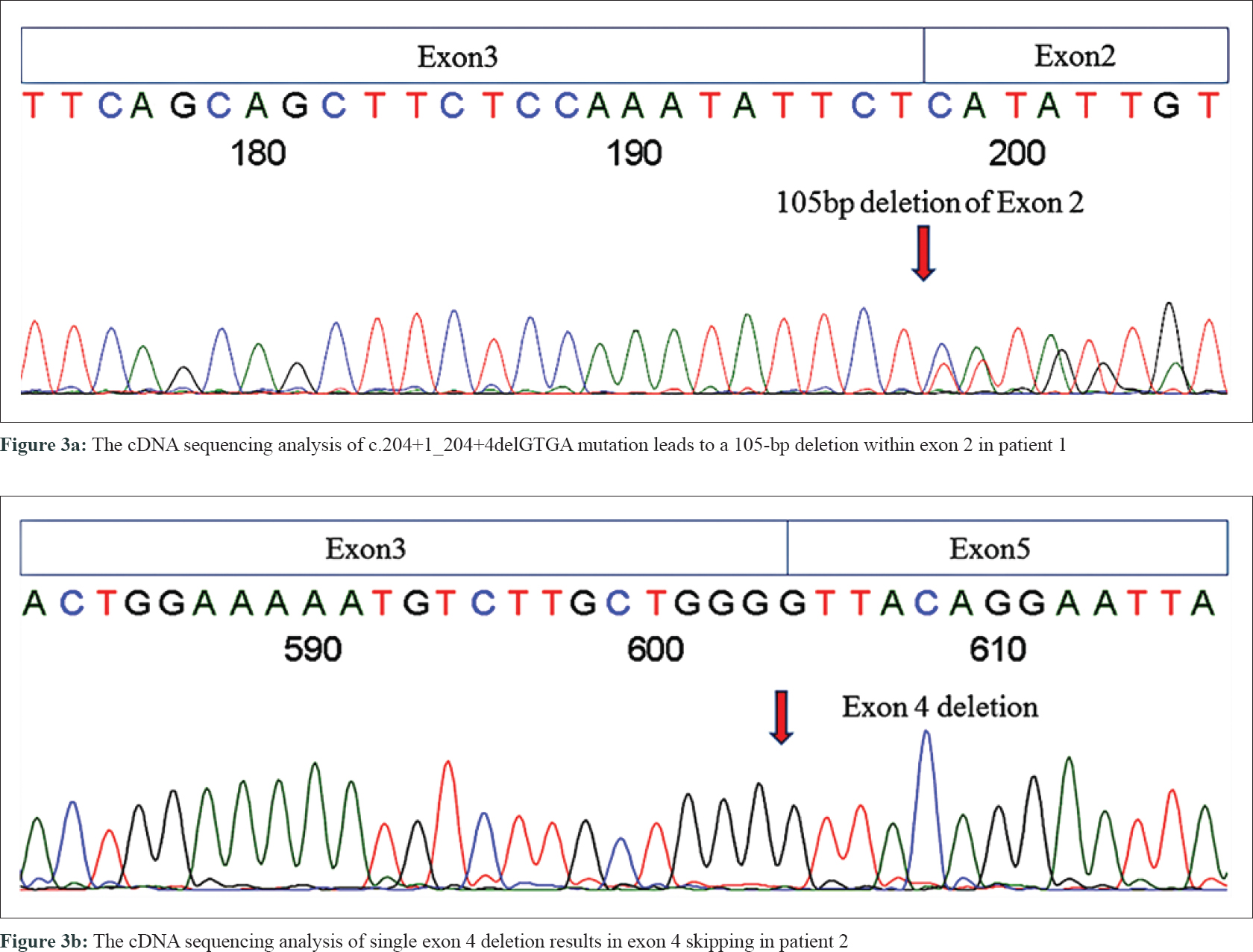
In the proband of family 1 (patient 1), a novel splice-site 4-bp deletion (c. 204 + 1_204 + 4delGTGA) was detected in intron 2, which caused a partial skipping of exon 2. This created an abnormal messenger RNA (mRNA) transcript containing a deletion of 105 nucleotides of exon 2, as confirmed by cDNA sequencing analysis [Figure - 3]a. Translation of this abnormal mRNA transcript would produce the substitution of valine with arginine at amino acid position 34 (p. Val34Arg) and an in-frame deletion of 35 amino acids between positions 33 and 69. Another novel mutation is a single-exon deletion of exon 4, that was identified by MLPA analysis in the proband of family 2 (patient 2). The single exon 4 deletion was also confirmed by cDNA sequencing to result in an aberrant mRNA transcript containing a deletion of 191 nucleotides between exons 3 and 5 [Figure - 3]b, which would produce an NF1 protein with the substitution of glutamine with valine at amino acid position 97 (p. Gln97Val) and a frameshift deletion of 63 amino acids (from amino acid positions 96 to 160). The frameshift deletion introduces a stop codon (TGA) at amino acid position 109. This results in a truncated protein with only 108 amino acids, that is far smaller than normal neurofibromin (2818 AAs). Neurofibromin has two major functional domains. One is the GTPase-activating protein domain, which is encoded by exons 21–27. The other one is cyclic adenosine monophosphate–dependent protein kinase recognition sites, encoded by exons 11–17.[5] The splice-site mutation in patient 1 produced an abnormal protein with all the functional domains. The exon 4 deletion in patient 2 resulted in a truncated short peptide without any functional domains, This resulted in the functional deficiency of neurofibromin (due to the loss of one copy of NF1). We also found two previously reported mutations at splice sites c.1527+1G>A (in family 3 case, patient 3) and c.6641+1G>A (in the sporadic case, patient 4).[6],[7] Pathogenicity analyses by MutationTaster indicated that the two current variants were both deleterious.[8] These mutations were all co-segregated with NF1 phenotype in the three families, carried by all the related patients and absent from unaffected individuals [Table - 3]. None of these mutations were found in 100 unrelated controls and in dbSNP database.[9]
 |
| Figure 3: |
More than 2600 different mutations, ranging from single-nucleotide substitutions to large deletions, have been reported in NF1 patients according to Human Gene Mutation Database.[10] It appears that the mutations are distributed throughout the entire NF1 gene without any hotspots.[11] To date, only a few unambiguous genotype–phenotype correlations of NF1 have been identified. Alkindy et al. found that patients with splice-site mutation have a significantly increased risk of developing neoplasm, particularly malignant peripheral nerve sheath tumors and central nerve system gliomas, compared with those with other mutations in NF1 gene.[12] In this study, all the four patients showed severe clinical manifestations of NF1 phenotype, including a large number of cutaneous and subcutaneous neurofibromas and huge neoplasm on the trunk. However, the three patients with splice-site mutations (patients 1, 3, and 4) did not suffer from malignancy. In addition, patient 1 also showed calvarial defect, which may be due to the abnormal protein with all the functional domains, produced by the splice-site mutation, that may interfere the function of normal protein. Patient 2 did not suffer from learning disability, bony dysplasia, or any malignancy, which could be the effect of the functional insufficiency of neurofibromin. However, due to the limited number of patients analyzed in our study, these phenotype–genotype correlations need to be confirmed by further studies.
In summary, our study discovered two novel mutations of NF1 gene that cause either abnormal functionality or functional deficiency of neurofibromin. We also found a case of congenital skull defect in a NF1 patient with abnormal function of neurofibromin. Splice-site mutations were not found to be associated with malignancy, or related phenotypes. These results have helped to further expand the NF1 mutation database, apart from providing clinically useful information for genetic counselling and prenatal diagnosis of NF1.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patients have given their consent for their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Acknowledgement
The authors are most grateful to all the individuals who participated in this study. They would also like to thank Prof. Ming Li for his assistance in experiments and data analysis.
Financial support and sponsorship
This work was funded by the National Natural Science Foundation of China (81573040 and 81872520).
Conflicts of interest
There are no conflicts of interest.
| 1. |
Evans DG, O'Hara C, Wilding A, Ingham SL, Howard E, Dawson J, et al. Mortality in neurofibromatosis 1: In North West England: An assessment of actuarial survival in a region of the UK since 1989. Eur J Hum Genet 2011;19:1187-91.
[Google Scholar]
|
| 2. |
Hinman MN, Sharma A, Luo G, Lou H. Neurofibromatosis type 1 alternative splicing is a key regulator of Ras signaling in neurons. Mol Cell Biol 2014;34:2188-97.
[Google Scholar]
|
| 3. |
Rasmussen SA, Friedman JM. NF1 gene and neurofibromatosis 1. Am J Epidemiol 2000;151:33-40.
[Google Scholar]
|
| 4. |
Neurofibromatosis. Conference statement. National Institutes of Health consensus development conference. Arch Neurol 1988;45:575-8.
[Google Scholar]
|
| 5. |
Trovó-Marqui AB, Tajara EH. Neurofibromin: A general outlook. Clin Genet 2006;70:1-13.
[Google Scholar]
|
| 6. |
De Luca A, Schirinzi A, Buccino A, Bottillo I, Sinibaldi L, Torrente I, et al. Novel and recurrent mutations in the NF1 gene in Italian patients with neurofibromatosis type 1. Hum Mutat 2004;23:629.
[Google Scholar]
|
| 7. |
Pros E, Gómez C, Martín T, Fábregas P, Serra E, Lázaro C. Nature and mRNA effect of 282 different NF1 point mutations: Focus on splicing alterations. Hum Mutat 2008;29:E173-93.
[Google Scholar]
|
| 8. |
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat Methods 2014;11:361-2.
[Google Scholar]
|
| 9. |
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. DbSNP: The NCBI database of genetic variation. Nucleic Acids Res 2001;29:308-11.
[Google Scholar]
|
| 10. |
Stenson PD, Mort M, Ball EV, Evans K, Hayden M, Heywood S, et al. The human gene mutation database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum Genet 2017;136:665-77.
[Google Scholar]
|
| 11. |
Zhang J, Tong H, Fu X, Zhang Y, Liu J, Cheng R, et al. Molecular characterization of NF1 and neurofibromatosis type 1 genotype-phenotype correlations in a Chinese population. Sci Rep 2015;5:11291.
[Google Scholar]
|
| 12. |
Alkindy A, Chuzhanova N, Kini U, Cooper DN, Upadhyaya M. Genotype-phenotype associations in neurofibromatosis type 1 (NF1): An increased risk of tumor complications in patients with NF1 splice-site mutations? Hum Genomics 2012;6:12.
[Google Scholar]
|
Fulltext Views
9,500
PDF downloads
1,507





![[Table - 1]](#tbl_ijdvl_2020_86_1_76_272866_t4.jpg){kind=link}
![[Figure - 1]](#fig_ijdvl_2020_86_1_76_272866_f1.jpg){kind=link}
![[Table - 2]](#tbl_ijdvl_2020_86_1_76_272866_t5.jpg){kind=link}
![[Table - 3]](#tbl_ijdvl_2020_86_1_76_272866_t6.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2020_86_1_76_272866_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2020_86_1_76_272866_f3.jpg){kind=link}