Translate this page into:
Painful leg rash
2 Department of Dermatology, Changi General Hospital, Singapore
Correspondence Address:
Yee-Leng Teoh
Department of Dermatology, Changi General Hospital, 2 Simei Street 3, 529889.
Singapore
| How to cite this article: Yong AM, Teoh YL, Tay YK. Painful leg rash. Indian J Dermatol Venereol Leprol 2019;85:129 |
A 49-year-old woman presented at the Changi General Hospital, Singapore with an 18-month history of leg rashes associated with numbness. There was no fever or associated constitutional symptoms. She underwent hysterectomy for uterovaginal prolapse a year earlier. Cutaneous examination revealed brown, mildly tender nodules on the shins with livedo racemosa [Figure - 1]a and [Figure - 1]b. Laboratory results showed an elevated erythrocyte sedimentary rate of 35 mmol/h (normal range: 3–15 mm/h). The complete blood count, creatinine, liver function test and chest radiograph were normal. Hepatitis B and C screen, antineutrophil cytoplasmic antibodies and antinuclear antibodies were negative. Antithrombin III, Factor V Leiden, protein C and S, extractable nuclear antigen screen, β-2 microglobulin, anticardiolipin antibodies and homocysteine levels were normal. A skin biopsy from her shin showed focal fibrinoid necrosis of medium-sized blood vessels in the subcutis with a surrounding dermal lymphohistiocytic infiltrate. There was thrombosis of the vessel lumen. The epidermis was unremarkable [Figure - 2]a and [Figure - 2]b. A review of the histology from the cervix specimen demonstrated necrotizing arteritis with fibrinoid necrosis and perivascular inflammatory cell infiltration [Figure - 3].
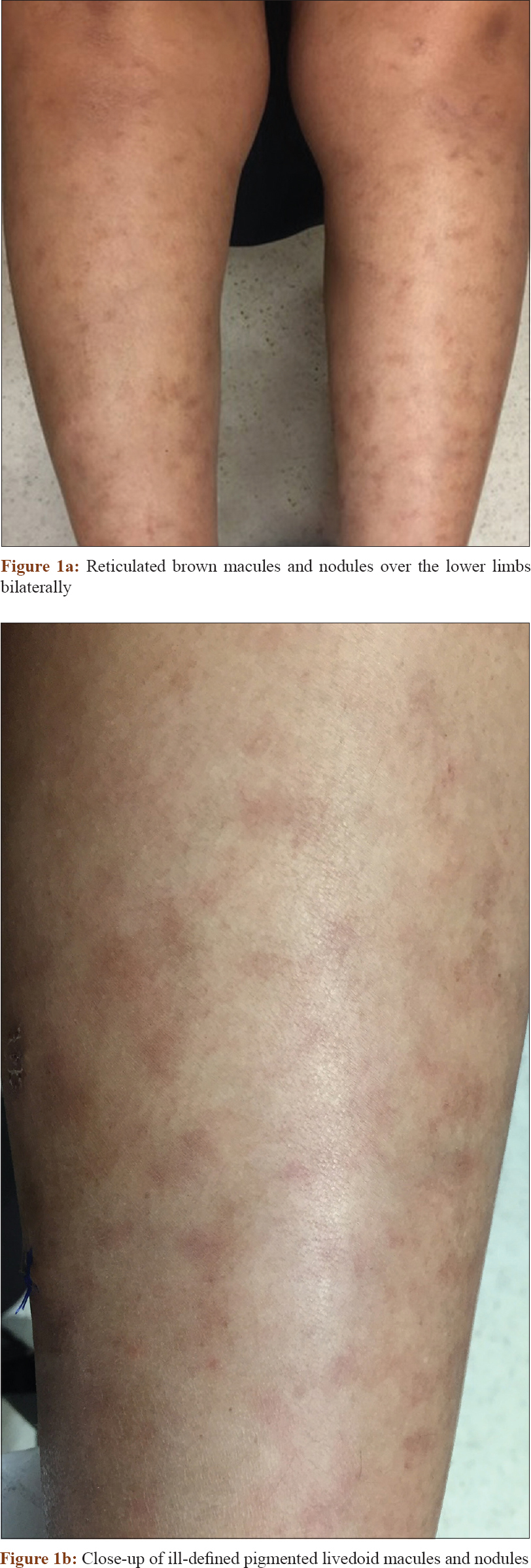 |
| Figure 1 |
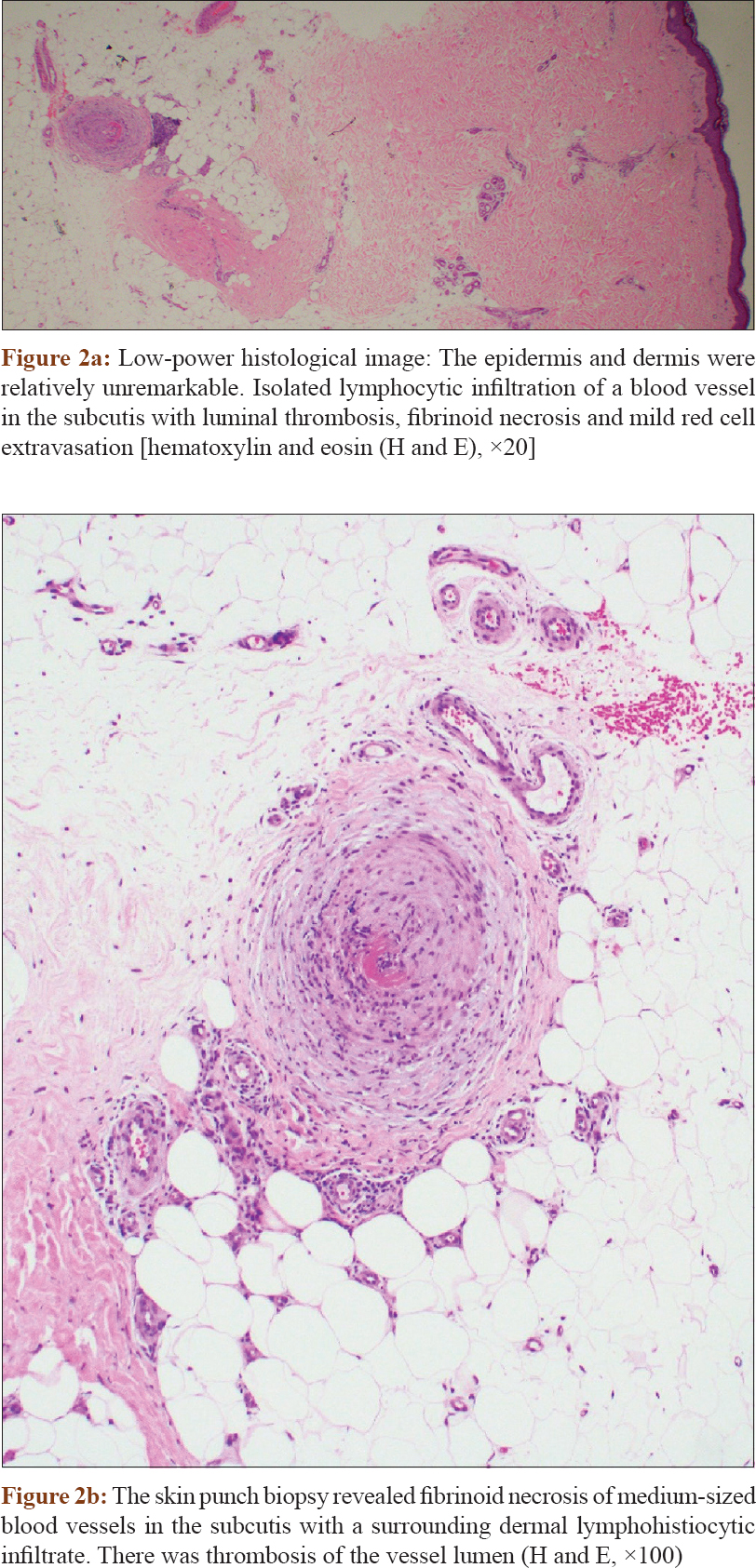 |
| Figure 2 |
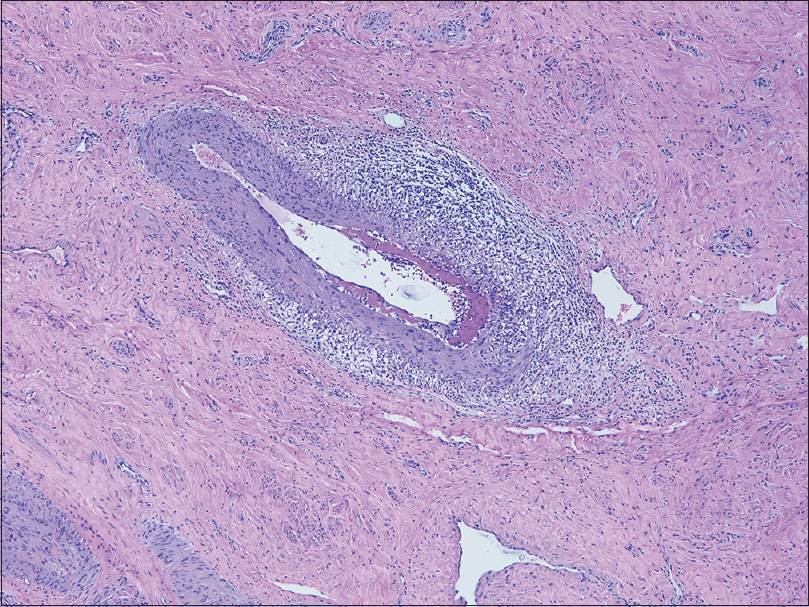 |
| Figure 3: The cervical specimen biopsy demonstrates fibrinoid necrosis of a medium-sized vessel wall in the dermis and a lymphocyte-predominant perivascular inflammatory cell infiltration (H and E, ×100) |
What Is the Diagnosis?
Answer
Polyarteritis nodosa.
Discussion
Polyarteritis nodosa is a systemic vasculitis characterized by necrotizing inflammation affecting medium-sized and small muscular arteries. In view of livedo racemosa, neurological involvement and negative antineutrophil cytoplasmic antibodies, this is the most likely diagnosis. The American College of Rheumatology has established 10 criteria for the classification of polyarteritis nodosa.[1] Three of the 10 criteria showed a sensitivity of 82.2% and specificity of 86.6% for the classification of polyarteritis nodosa in a patient with a documented vasculitis [Table - 1].

The differential diagnoses include eosinophilic granulomatosis with polyangiitis, microscopic polyangiitis, granulomatosis with polyangiitis and Henoch-Schönlein purpura. Eosinophilic granulomatosis with polyangiitis is an eosinophilic-rich necrotizing vasculitis affecting small- to medium-sized vessels. Patients often have rhinosinusitis, asthma and blood eosinophilia which were not present in our patient. Microscopic polyangiitis is a pauci-immune necrotizing vasculitis affecting small vessels with variable involvement of medium-sized arteries. Antineutrophil cytoplasmic antibodies are present in more than 90% of patients. Granulomatosis with polyangiitis presents with nasal inflammation, pulmonary infiltrates or hemorrhage and hematuria. A negative antineutrophil cytoplasmic antibody and absence of necrotizing granulomatous inflammation on skin biopsy makes this condition an unlikely diagnosis for our patient. On the contrary, Henoch-Schönlein purpura is associated with palpable purpura, arthritis, abdominal pain and renal disease which were not observed in our case.
The risk of long-term systemic vasculitis in patients who have incidental single-organ vasculitis is low. However, if patients develop signs of systemic polyarteritis nodosa or giant cell arteritis, a thorough systemic evaluation should be considered. Isolated vasculitis of the female genital tract is rare, typically occurring in women in their 40s and often an incidental finding from hysterectomy specimens. Vasculitis of the genital female tract of the classical polyarteritis nodosa-type occurred in 91.5% of cases reported in the literature, and the rest presented with isolated giant cell arteritis-type. The polyarteritis nodosa-type commonly involved the cervix in up to 65.7%.[2]
Polyarteritis nodosa of the genitalia is a rare entity. The female genitalia tract is more commonly affected in isolated polyarteritis nodosa than the systemic form.[3] In addition, patients with systemic polyarteritis nodosa are older than patients with isolated polyarteritis nodosa. Isolated genitalia polyarteritis nodosa presents more frequently with vaginal bleeding and is rarely associated with asymptomatic pelvic masses. Moreover, when the condition is found in the female genital tract, isolated polyarteritis nodosa is primarily found in the cervix, whereas systemic polyarteritis nodosa involves multifocal aspects of the female genital tract.
In polyarteritis nodosa, the extent of organ involvement and disease progression guides treatment. The five-factor score revised in 2011 by the French Vasculitis Study Group showed that prognostic factors which predicted mortality include renal insufficiency (serum creatinine >1.58 mg/dL), proteinuria >1 g/day, gastrointestinal involvement, cardiomyopathy and age above 65 years.[4] Treatment mostly relies on corticosteroid therapy. In this patient with mild disease (five-factor score = 0), corticosteroids should be the initial treatment option of choice. First-line corticosteroid therapy is able to achieve and maintain remission in about 50% of patients with mild non-hepatitis B- and C-related polyarteritis nodosa. In contrast, other forms of therapy including cyclophosphamide, rituximab and plasma exchange have been shown to be effective in non-hepatitis B- and C-related polyarteritis nodosa; however, the usage is mainly reserved for those with frequent relapses involving multiple organs which are refractory to systemic steroid therapy.[5],[6] Our patient was treated with prednisolone 30 mg daily (0.5 mg/kg/day) for 2 weeks, followed by prednisolone 60 mg daily (1 mg/kg/day) for further 4 weeks. Her leg numbness and rashes improved, but she was subsequently lost to follow-up.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient has given her consent for her images and other clinical information to be reported in the journal. The patient understands that name and initials will not be published and due efforts will be made to conceal identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
| 1. |
Lightfoot RW Jr., Michel BA, Bloch DA, Hunder GG, Zvaifler NJ, McShane DJ, et al. The American College of Rheumatology 1990 criteria for the classification of polyarteritis nodosa. Arthritis Rheum 1990;33:1088-93.
[Google Scholar]
|
| 2. |
Hoppé E, de Ybarlucéa LR, Collet J, Dupont J, Fabiani B, Puéchal X. Isolated vasculitis of the female genital tract: A case series and review of literature. Virchows Arch 2007;451:1083-9.
[Google Scholar]
|
| 3. |
Hernández-Rodríguez J, Tan CD, Rodríguez ER, Hoffman GS. Gynecologic vasculitis: An analysis of 163 patients. Medicine (Baltimore) 2009;88:169-81.
[Google Scholar]
|
| 4. |
Guillevin L, Pagnoux C, Seror R, Mahr A, Mouthon L, Le Toumelin P, et al. The five-factor score revisited: Assessment of prognoses of systemic necrotizing vasculitides based on the French vasculitis study group (FVSG) cohort. Medicine (Baltimore) 2011;90:19-27.
[Google Scholar]
|
| 5. |
de Luna G, Chauveau D, Aniort J, Carron PL, Gobert P, Karras A, et al. Plasma exchanges for the treatment of severe systemic necrotizing vasculitides in clinical daily practice: Data from the French vasculitis study group. J Autoimmun 2015;65:49-55.
[Google Scholar]
|
| 6. |
Seri Y, Shoda H, Hanata N, Nagafuchi Y, Sumitomo S, Fujio K, et al. Acase of refractory polyarteritis nodosa successfully treated with rituximab. Mod Rheumatol 2017;27:696-8.
[Google Scholar]
|
Fulltext Views
3,932
PDF downloads
3,389





![[Figure - 1]](#fig_ijdvl_2019_85_1_129_246743_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2019_85_1_129_246743_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2019_85_1_129_246743_f3.jpg){kind=link}
![[Table - 1]](#tbl_ijdvl_2019_85_1_129_246743_t4.jpg){kind=link}