Translate this page into:
Novel transglutaminase 1 mutations in a Chinese patient with severe lamellar ichthyosis phenotype
Correspondence Address:
Hong Fang
Department of Dermatology, The First Affiliated Hospital, College of Medicine, Zhejiang University, No. 79, Qingchun Road, Hangzhou, Zhejiang Province
People's Republic of China
| How to cite this article: Bai J, Ding Yg, Wu Yh, Qiao Jj, Fang H. Novel transglutaminase 1 mutations in a Chinese patient with severe lamellar ichthyosis phenotype. Indian J Dermatol Venereol Leprol 2015;81:292-294 |
Sir,
Autosomal recessive congenital ichthyosis (ARCI) has three clinical subtypes: lamellar ichthyosis (LI; OMIM 242300), congenital ichthyosiform erythroderma (CIE; OMIM 242100), and harlequin ichthyosis (HI; OMIM 242500). [1] Lamellar ichthyosis is a rare, autosomal recessive skin disorder characterized by generalized hyperkeratosis, with estimated incidence rates of 1:250000. [2] Genetic mutations reported to cause lamellar ichthyosis include mutations in transglutaminase 1 (TGM1), ABCA12, CGI-58/ABHD5, FLJ39501 (a homolog of CYP4F2), ALOX12B, ALOXE-3, and ichthyin (NIPAL4). [3] This report describes the identification of a novel mutation of the TGM1 gene in a Chinese patient with the phenotype of severe lamellar ichthyosis.
The proband was a 42-year-old Chinese man who presented with generalized, large, gray-brown, plate-like scales covering the entire body surface including the face and flexures. Though not in frank erythroderma, he had universal alopecia, severe ectropion of eyelids, conjunctival congestion, eclabium, palmoplantar hyperkeratosis with fissuring and thickening and dystrophy of the nail plates which resembled glossy spoons. He also had severe hypohidrosis, dysplasia of the auricular cartilage, teeth abnormalities, and deformities of the small hand joints. His left eye had impaired vision due to corneal ulceration. History of consanguinity was present; his parents were second cousins. His mother had died of hepatic carcinoma in 2009. The parents had normal skin but all his siblings had a skin problem similar to his and had died shortly after birth, possibly due to infections [Figure - 1]. Light microscopy of lesional skin showed marked hyperkeratosis and acanthosis with a normal granular layer. Neutrophil collections were found beneath the stratum corneum [Figure - 2]i-j. Based on the clinical and histopathological features, a diagnosis of lamellar ichthyosis was made.
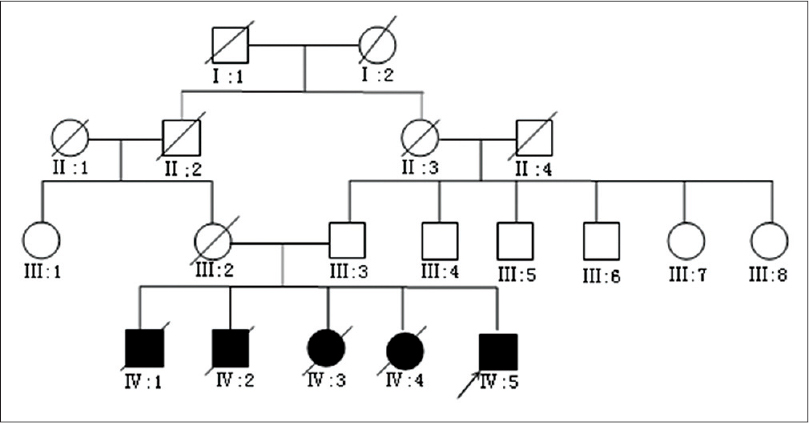 |
| Figure 1: Pedigrees of the family. His parents are consanguineous and his brothers and sisters died shortly after birth |
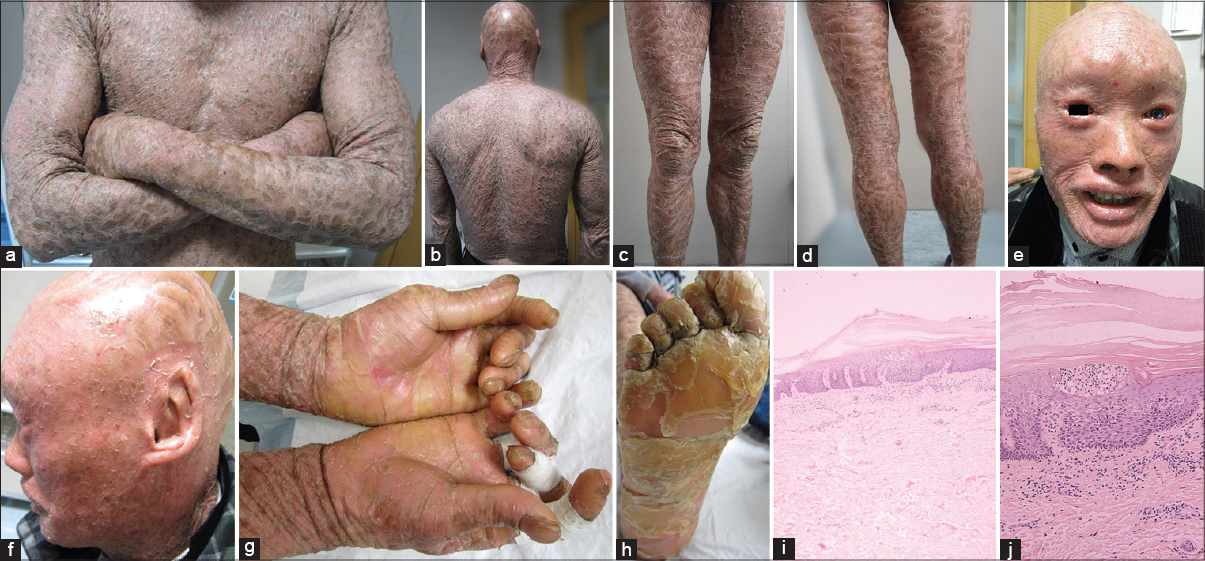 |
| Figure 2: The patient shows (a-d) generalized large gray or brownish lamellar scales (e) alopecia, ectropion, eclabium, teeth abnormalities, corneal opacity of left eye, (f) dysplasia of auricular cartilage, (g, h) palmoplantar hyperkeratosis and deformities of small hand joints, (i) marked hyperkeratosis with a follicular plug, a normal granular layer and acanthosis (H and E, ×20), (j) Infiltration of neutrophils (H and E, ×100) |
After obtaining informed consent from all participants, DNA was extracted from the peripheral blood of the patient, his father and unrelated controls. The exon sequences of transglutaminase1 gene (TGM1) were amplified by polymerase chain reaction (PCR) using an automated sequencing system. A novel missense mutation was found. The patient′s DNA harboured a homozygous mutation of A → C transition located in exon 3(c. 386A > C) of TGM1, resulting in the substitution of histidine for proline at amino acid position 129(p.H129P). In a three-dimensional model of human transglutaminase 1, it appeared at the beginning of the N-terminal beta-sandwich domain [Figure - 3]. The presence of the mutation was confirmed with re-sequencing of repeated PCR products. It was not found in 50 samples from unrelated healthy controls. The patient′s father′s DNA was heterozygous for the above-mentioned p.H129P mutation, which further supports the likelihood of its pathogenic role. As his parents were consanguineous, we speculate that the patient inherited the p.H129P mutation from both his parents. However, his mother′s sample was not available to confirm our theory. We conclude that p.H129P (c. 386A > C) is a novel mutation of TGM1 responsible for lamellar ichthyosis; we were unable to find any previous reports of this mutation in the available literature and it was also not found in any of the 50 healthy controls.
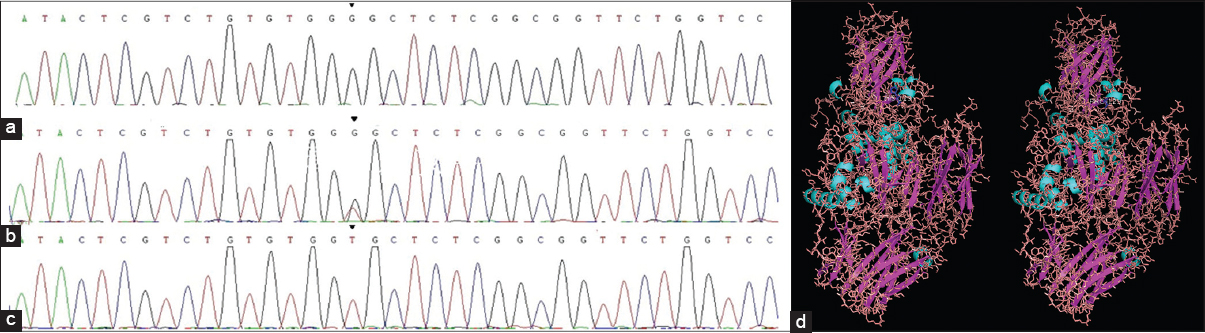 |
| Figure 3: Mutations in the TGM1 gene and protein (a) homozygous c.386A>C mutation in exon3 and (b) the heterozygous mutation of his father (c) normal control sequence (d) 3D image of human transglutaminase 1 showing the substitution of His 129 for Pro 129 (blue) |
H129P codes for the same domain of TGM1 which is affected by the commonly reported R143C mutation in patients with lamellar ichthyosis. We speculate that the H129P mutation reduces enzyme stability, just as R143C does. A mouse line with only the R142C point mutation (corresponding to R143C in humans) demonstrated low levels of the mutant TGM1 protein in the skin with loss of enzyme activity, confirming that the phenotype of Tgm1 R142C/R142C mice becomes comparable to that of Tgm1 -/- mice. [4] By extrapolation, it is understandable why our patient has a severe phenotype. A mouse model with this novel point mutation will be useful to elucidate the correlation between phenotype and genotype accurately, and will also aid the development of new therapeutic strategies.
Patients diagnosed with lamellar ichthyosis need lifelong treatment. Three important principles are involved in treatment: hydration, lubrication, and keratolysis. Topical emollients, keratolytic agents and other medications that are employed include urea, salicylic acid, bath oils, tazarotene N-acetylcysteine calcipotriol and tacrolimus; in more severe cases, oral retinoids are suggested. [5] Our patient was treated with urea cream, salicylic acid cream, adapalene gel, chlortetracycline eye ointment and oral vitamin A (25000 IU bid). His clinical features have improved and he follows up regularly.
In conclusion, our work expands the TGM1 mutation spectrum. Definitive molecular diagnosis for individuals at risk of producing an affected child, obtained by mutation screening, can not only provide more accurate prenatal genetic counseling, but also, hopefully, lead to new treatments.
| 1. |
Louhichi N, Hadjsalem I, Marrakchi S, Trabelsi F, Masmoudi A, Turki H, et al. Congenital lamellar ichthyosis in Tunisia is caused by a founder nonsense mutation in the TGM1 gene. Mol Biol Rep 2013;40:2527-32.
[Google Scholar]
|
| 2. |
Huber M, Yee VC, Burri N, Vikerfors E, Lavrijsen AP, Paller AS, et al. Consequences of seven novel mutations on the expression and structure of keratinocyte transglutaminase. J Biol Chem 1997;272:21018-26.
[Google Scholar]
|
| 3. |
Terrinoni A, Serra V, Codispoti A, Talamonti E, Bui L, Palombo R, et al. Novel transglutaminase 1 mutations in patients affected by lamellar ichthyosis. Cell Death Dis 2012;3:e416.
[Google Scholar]
|
| 4. |
Nakagawa N, Yamamoto M, Imai Y, Sakaguchi Y, Takizawa T, Ohta N, et al. Knocking-in the R142C mutation in transglutaminase 1 disrupts the stratum corneum barrier and postnatal survival of mice. J Dermatol Sci 2012;65:196-206.
[Google Scholar]
|
| 5. |
Vahlquist A, Ganemo A, Virtanen M. Congenital ichthyosis: An overview of current and emerging therapies. Acta Derm Venereol 2008;88:4-14.
[Google Scholar]
|
Fulltext Views
3,410
PDF downloads
1,407





![[Figure - 1]](#fig_ijdvl_2015_81_3_292_154786_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2015_81_3_292_154786_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2015_81_3_292_154786_f3.jpg){kind=link}