Translate this page into:
Bullous pemphigoid clinically presenting as lichen amyloidosis
2 Department of Dermatology, Radha Gobinda Kar Medical College, Kolkata, West Bengal, India
Correspondence Address:
Projna Biswas
Depatrment of Dermatology, Institute of Post Graduate Medical Education and Research, Kusumba (SDPark), PO Narendrapur, PS Sonarpur, Kolkata - 700 103, West Bengal
India
| How to cite this article: Biswas P, Aggarwal I, Sen D, Sumi A, Ghosh A. Bullous pemphigoid clinically presenting as lichen amyloidosis . Indian J Dermatol Venereol Leprol 2014;80:544-546 |
Sir,
A 42-year-old male presented to our hospital with complaints of moderately itchy, pigmented, papular lesions over the lower limbs for the last 2 years. There was mild itching to start with, following which the patient noticed gradual appearance of papules over his shins. There was no preceding history of any drug intake. On examination, there were multiple, discrete, closely-set skin-colored and pigmented papules of varying sizes symmetrically present over the antero-lateral aspect and calves of lower limbs symmetrically [Figure - 1]. There was a sharp demarcation line between the lesions and normal skin. Individual papules were of about 1 cm in diameter, non-tender with mild scaling but without oozing or excoriation. No other body site including mucosae, scalp, or nails was involved. We clinically suspected the case to be lichen amyloidosis and skin biopsy was sent with a request to the pathologist for a Congo red stain.
Histopathological examination (HPE) revealed marked hyperkeratosis, normal to thinned-out stratum malphigii, mild spongiosis along with a subepidermal bulla. There was a considerable collection of eosinophils, both in the blister cavity and in the upper dermis along with neutrophils and lymphocytes [Figure - 2]. There was no evidence of any amyloid deposits with Congo red stain. These features were suggestive of bullous pemphigoid and subsequent perilesional skin biopsy for direct immunofluorescentce (DIF) examination showed deposition of IgG and C3 in the basement membrane zone [Figure - 3]. Since topical clobetasol propionate, 0.05% ointment did not reduce the lesion count and size, we treated the patient with oral prednisolone 60 mg/day. The lesions regressed, but on gradual tapering to 20 mg of prednisolone per day over 8 weeks, the lesions stared recurring again. At this point, we lost the patient to follow up.
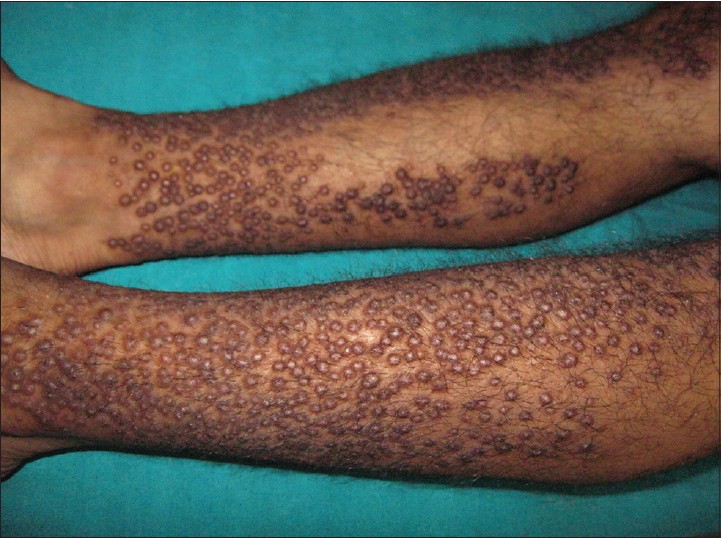 |
| Figure 1: Multiple discrete close-set papules in lower limb |
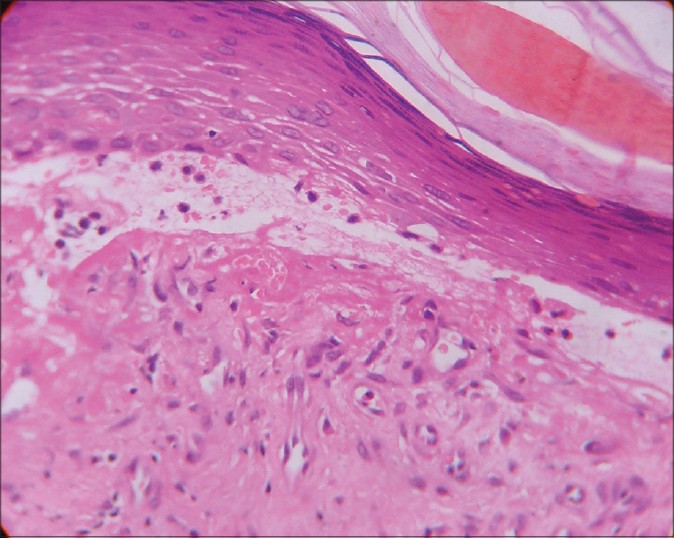 |
| Figure 2: Subepidermal blister with upper dermal eosinophil and lymphocyte collection within the blister cavity (H and E, x400) |
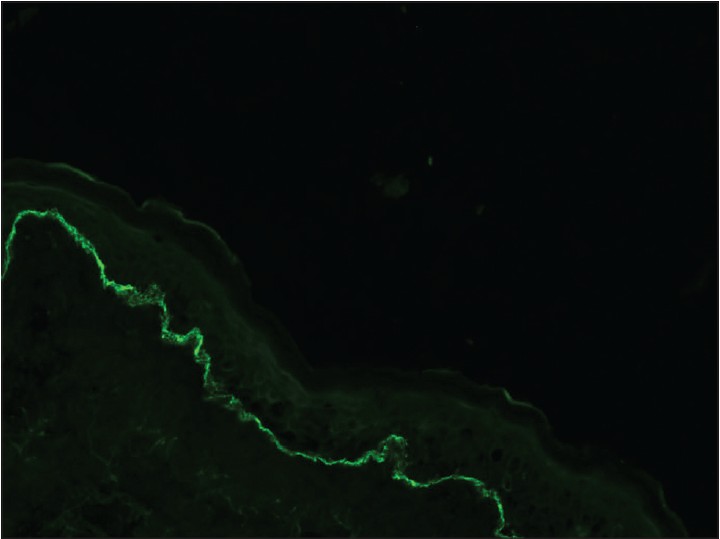 |
| Figure 3: Direct immunoflourescence showing presence of fine, linear continuous deposits along the epidermal basement membrane (x100) |
Bullous pemphigoid (BP) is the most common autoimmune subepidermal blistering disorder occurring in elderly individuals >60 years of age. There is tissue-bound and circulating autoantibody against BP230 and BP180. In addition, there is an autoreactive cell-mediated immune response against BP180. [1] Histopathology shows a subepidermal cleft with eosinophilic spongiosis and eosinophilic and mononuclear cell infiltrates in the upper dermis; direct immunoflourescence demonstrates presence of fine, linear continuous deposits of IgG and/or C3 along the epidermal basement membrane. Several clinical variants of bullous pemphigoid have been described including localized (such as pretibial pemphigoid, around stomas, vulvar and perineal region in female, at the site of irradiation or confined to paralyzed limb), isolated palmo-plantar involvement (dyshidrosiform pemphigoid), dermatitis herpetiformis-like (vesicular pemphigoid) erythrodermic bullous pemphigoid, lichen planus pemphigoides, gestational pemphigoid [2] and the rare pemphigoid incipiens (elderly patients with itchy BP like lesions but a negative DIF in the presence of circulating anti-BP180 and/or BP230 antibodies). [3] Prurigo nodularis-like pemphigoid (nodular pemphigoid) is a variant where elderly women present with excoriated papules and nodules distributed predominantly over lower limbs and shoulders. Blisters may precede or follow the development of nodules by many months, at the site of the nodule or on uninvolved skin. [4],[5] Since the lesions were very close-set and uniformly distributed in our patient, we did not consider pemphigoid nodularis as a possible differential diagnosis. The marked acanthosis and elongation of rete ridges with corresponding changes in the papillary dermis observed in pemphigoid nodularis were also not seen in our patient. Epidermolysis bullosa (EB) pruriginosa, a rare variant of dystrophic EB, may be considered in the differential diagnosi as a similar late presentation with subepidermal blisters and inflammatory infiltrate including eosinophils have been reported. [6] Lack of past history of trauma-induced blistering, absence of a family history, lack of involvement of nails and a positive DIF helped to differentiate our case from EB pruriginosa. However, we could not carry out an electron microscopy examination, nor could we demonstrate any missense mutation in COL 7A1 gene (described in EB pruriginosa) as we did not have access to these investigations at our institute. [7] The age of onset of disease in our patient was much less than the usual for bullous pemphigoid; in addition, he did not develop clinical vesicles or bullae even after 2 years of onset of the lesions. He had closely-set papules uniformly distributed only over the lower limbs, resembling the papules of cutaneous amyloidosis. A single case has been reported in the literature as ′lichen amyloidosis with subepidermal blister′ where the patient presented with multiple itchy keratotic papules and plaques on trunk and extremities with intermingled erosions and vesicles. The case was clinically reminiscent of prurigo nodularis but histopathology showed subepidermal bulla with amyloid deposit in the dermal papillae, visible on Dylon and Congo red staining. Direct immunofluorescence was normal. [8] In contrast, our case did not demonstrate any amyloid deposit in the dermal papillae.
| 1. |
Culton DA, Liu Z, Diaz LA. Bullous pemphigoid.In: Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ, Editors. Fitzpatrick's Dermatology in General Medicine. 8 th ed.New York: McGraw-Hill; 2012.p. 608-16.
th ed.New York: McGraw-Hill; 2012.p. 608-16.'>[Google Scholar]
|
| 2. |
Borradori L, BernardP. Pemphigoidgroup. In: Bolognia JL, Jorizzo JL, Rapini RP, editors. Dermatology. 2 nd ed. Spain: Mosby Elsevier; 2008.p. 137-48.
[Google Scholar]
|
| 3. |
Tambe S, Häfliger S, Borradori L. Clinical challenges and recent advances in the diagnosis of bullous pemphigoid. Expert Rev Dermatol 2013;8:407-16.
[Google Scholar]
|
| 4. |
Borradori L, Prost C, Wolkenstein P, Bernard P, Baccard M, Morel P. Localized pretibialpemphigoid and pemphigoidnodularis. J Am Acad Dermatol 1992;27:863-7.
[Google Scholar]
|
| 5. |
Cliff S, Holden CA. Pemphigoidnodularis: A report of three cases and review of the literature. Br J Dermatol 1997;136:398-401.
[Google Scholar]
|
| 6. |
Vivehanantha S, Carr RA, McGrath JA, Taibjee SM, Madhogaria S, Ilchyshyn A. Epidermolysisbullosapruriginosa: A case with prominent histopathologic inflammation. JAMA Dermatol 2013;149:727-31.
[Google Scholar]
|
| 7. |
Shi BJ, Feng J. A novel missense mutation in the COL7A1 gene causes epidermolysisbullosapruriginosa. Clin Exp Dermatol 2009;34:e975-8.
[Google Scholar]
|
| 8. |
Kuroda K, Mizoguchi M. Lichen amyloidosus with subepidermal blister formation. Eur J Dermatol 2004;14:262-3.
[Google Scholar]
|
Fulltext Views
4,732
PDF downloads
1,509





![[Figure - 1]](#fig_ijdvl_2014_80_6_544_144184_u1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2014_80_6_544_144184_u2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2014_80_6_544_144184_u3.jpg){kind=link}