Translate this page into:
Vohwinkel syndrome with mental retardation
Correspondence Address:
P Mercy
Department of Dermatology and Venereology, Jawahar Lal Nehru Hospital and Research Centre, Bhilai - 490 009, Chhattisgarh
India
| How to cite this article: Mercy P, Singh A, Ghorpade A K, Das M N, Upadhyay A, Keswani N. Vohwinkel syndrome with mental retardation. Indian J Dermatol Venereol Leprol 2013;79:725 |
Sir,
Vohwinkel syndrome, known as keratoderma hereditaria mutilans, is a syndromic form of diffuse palmoplantar keratoderma (PPK) that manifests as hyperkeratosis of the palms and soles with a honeycomb appearance. It is a rare autosomal dominant palmoplantar keratoderma, which was first described in 1929 by Vohwinkel. Clinically, this condition manifests in infants, becomes transgredient during childhood. Constricting fibrous bands appear at later stages on the digits and can lead to progressive strangulation and auto amputation (pseudoainhum). Starfish-shaped keratosis may occur on the elbows, knees and the dorsa of the hands and feet, a characteristic feature of this disorder. Some associated features have been noted and include alopecia, hearing loss, spastic paraplegia, myopathy, ichthyosiform dermatoses, and nail abnormalities. [1],[2],[3] Recent molecular biological studies indicate the presence of two variants of Vohwinkel syndrome, an (1) ichthyosis-associated variant, associated with an insertional mutation of the loricrin gene, and a (2) deafness-associated variant, associated with a missense mutation of the connexin-26 gene. [2],[4]
Two siblings, aged 18 years and 12 years, born out of second degree consanguineous marriage with severe growth retardation presented with severe trans gradient honey comb like PPK with digitate projections over the normal skin. In both the siblings keratoderma started in early part of the life (<5 years) [Figure - 1]. Examination showed the presence of pseudoainhum with auto amputation of fingers and toes. Starfish keratosis was observed in both sisters over the dorsum of hands [Figure - 2]. Keratotic plaques were seen over both knees. Flexion contracture was also observed over fingers in both the sisters with tapering of the nails. There was a history of similar problem in their father and grandfather [Figure - 3]. Sensorineural deafness was also detected in both the sisters. Based on above mentioned characteristic findings we diagnosed Vohwinkel′s syndrome in these sisters. Mental retardation (I.Q. - 56 and 52) was also detected in both siblings. Patients were advised use of topical keratolytic in the form of 6% salicylic acid.
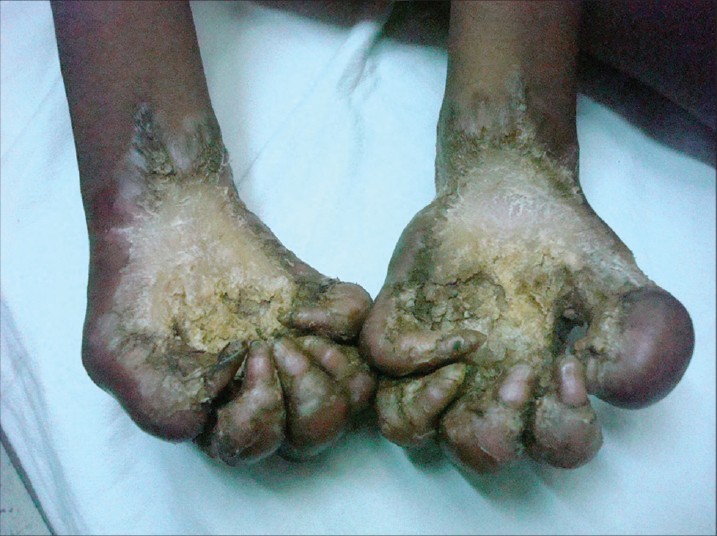 |
| Figure 1: Mutilating palmar keratoderma |
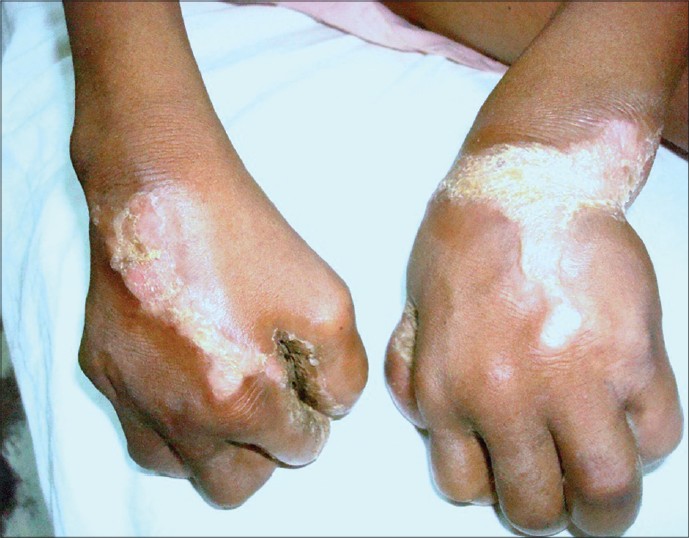 |
| Figure 2: Keratotic plaques on dorsa of hands |
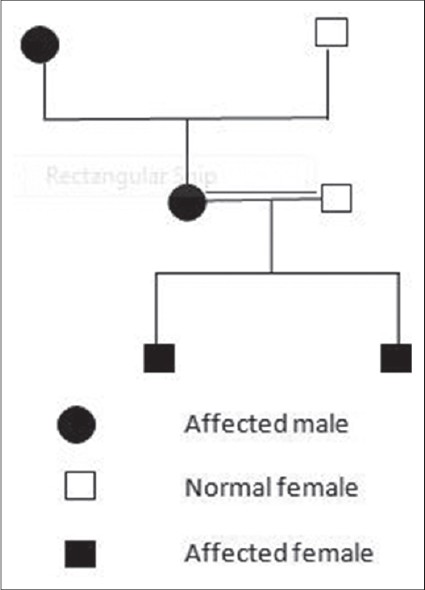 |
| Figure 3: Pedigree chart |
In 1929, Vohwinkel reported a 24-year-old woman with rapidly progressive PPK having reddish blue border and pseudoainhum of several digits. The condition was inherited in an autosomal dominant fashion. The diagnosis has largely been morphological, based on the classical triad of diffuse PPK in a honeycombed pattern, constricting bands of the digits with auto amputation (pseudoainhum), and starfish-shaped hyperkeratotic plaques on the dorsum of the hands and feet, elbows, and knees. Linear hyperkeratosis may also be observed on the elbows and knees. [1],[2],[3] Histopathological features of this syndrome include hyperkeratosis, parakeratosis, acanthosis and hypergranulosis.
The treatment of all types of hereditary keratoderma is difficult. It is mostly symptomatic and treatment may vary from simple measures, such as salt-water soaks, paring, topical keratolytic, systemic retinoid to reconstructive surgery, and total excision of the hyperkeratotic skin followed by grafting. The mainstays of treatment include topical keratolytic, topical retinoid and oral retinoid. Oral retinoid are effective, especially in some hereditary PPKs. [4] the prognosis is good as long as medications are used; patients may have a normal life span.
Differential diagnosis of Vohwinkel syndrome includes acral keratoderma and Olmsted syndrome. Acral keratoderma can be differentiated by the absence of star fish shaped keratosis, presence of striate hyperkeratosis of palms, and linear hyperkeratosis over acral areas. [5] Olmsted syndrome can be differentiated by the presence of mutilating PPK with periorificial hyperkeratosis.
We are reporting these cases because of very few reports in Indian literature with such severe form of mutilating keratoderma. Here we are reporting one additional feature in the form of mental retardation.
| 1. |
Lucker GP, Van de Kerkhof PC, Steijlen PM. The hereditary palmoplantarkeratoses: An updated review and classification. Br J Dermatol 1994;131:1-14.
[Google Scholar]
|
| 2. |
Itin PH, Fistarol SK. Palmoplantarkeratodermas. Clin Dermatol 2005;23:15-22.
[Google Scholar]
|
| 3. |
Gibbs RC, Frank SB. Keratomahereditariamutilans (Vohwinkel). Differentiating features of conditions with constriction of digits. Arch Dermatol 1966;94:619-25.
[Google Scholar]
|
| 4. |
Gedicke MM, Traupe H, Fischer B, Tinschert S, Hennies HC. Towards characterization of palmoplantarkeratoderma caused by gain-of-function mutation in loricrin: Analysis of a family and review of the literature. Br J Dermatol 2006;154:167-71.
[Google Scholar]
|
| 5. |
Rastogi S, Kumar P, Mukhija RD. Mutilating keratoderma with deaf-mutism. Indian J Dermatol Venereol Leprol 1996;62:254-5.
[Google Scholar]
|
Fulltext Views
2,855
PDF downloads
2,522





![[Figure - 1]](#fig_ijdvl_2013_79_5_725_116739_f1.jpg){kind=link}
![[Figure - 2]](#fig_ijdvl_2013_79_5_725_116739_f2.jpg){kind=link}
![[Figure - 3]](#fig_ijdvl_2013_79_5_725_116739_f3.jpg){kind=link}