
Translate this page into:
A novel KITLG mutation causes familial progressive hyperpigmentation and hypopigmentation with multiple café-au-lait macules

Corresponding author: Dr. Huijun Wang, Department of Paediatric Dermatology, Dermotology Hospital, Southern Medical University, Guangzhou, Guangdong, China. drhuijunwang@pku.edu.cn
-
Received: ,
Accepted: ,
How to cite this article: Huang X, Niu G, Lin Z, Wang H. A novel KITLG mutation causes familial progressive hyperpigmentation and hypopigmentation with multiple café-au-lait macules. Indian J Dermatol Venereol Leprol. doi: 10.25259/IJDVL_1246_2023
Dear Editor,
Familial progressive hyperpigmentation and hypopigmentation (FPHH, OMIM #145250), also known as melanosis universalis hereditaria, is an inherited skin disorder characterised by diffuse, progressive skin hyperpigmentation with hypopigmented spots. Additional features include multiple café-au-lait macules (CALMs), and lentiginosis intermingled with large hypopigmented ash-leaf macules.1 In 2009, the causative gene of FPHH has been identified as the KITLG gene which encodes the ligand for the KIT receptor tyrosine kinase.2 This case reports on a sporadic case of FPHH with a previously unknown KITLG variant.
A 15-year-old boy of Chinese Han ethnicity, born to healthy parents, presented with diffuse skin hyperpigmentation and multiple hypopigmented spots shortly after birth. As he grew older, CALMs and lentigines gradually appeared and increased in number. On examination, multiple diffuse lentigines were observed on his face, trunk, and extremities, accompanied by multiple CALMs and scattered hypopigmented macules of varying sizes [Figures 1a–1c]. Family history was non contributary. Based on the above clinical features, a diagnosis of FPHH was suspected. To find out the pathogenic variant, the patient’s peripheral blood DNA was subjected to whole-exome sequencing after informed consent. A heterozygous, missense variant c.113A>C (p.Lys38Thr) in KITLG was detected and further confirmed by direct sequencing [Figure 2a]. This variant was not detected in his parents indicating that it could be a de novo variant. In addition, this variant was absent in the public databases such as ExAC, dbSNP, and gnomAD. The affected residue Lys38 is located in a conserved β-strand of the KITLG protein. In silico analysis with the Mutation Taster software predicted the variant to be ‘disease causing’. According to the American College of Medical Genetics and Genomics (ACMG)/Association for Molecular Pathology (AMP) guidelines, it was classified as likely pathogenic. Otherwise, no suspected disease-causing variants in NF1 or SPRED1 leading to similar manifestations were identified in the whole-exome sequencing.
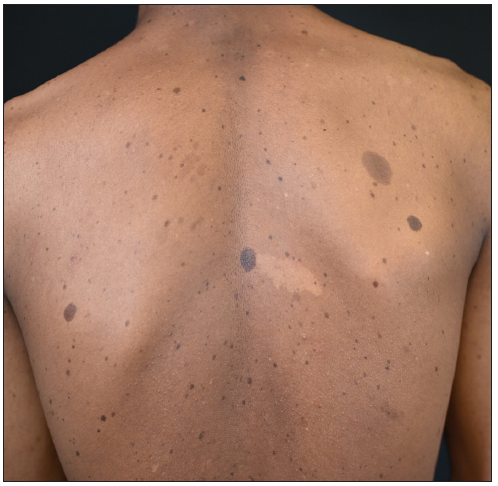
- Multiple lentigines with scattered café-au-lait macules and large hypopigmented ash-leaf macules distributed on the patient’s back.
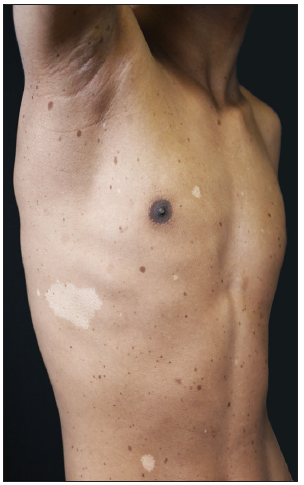
- Multiple lentigines with several hypopigmented macules on the patient’s right chest.
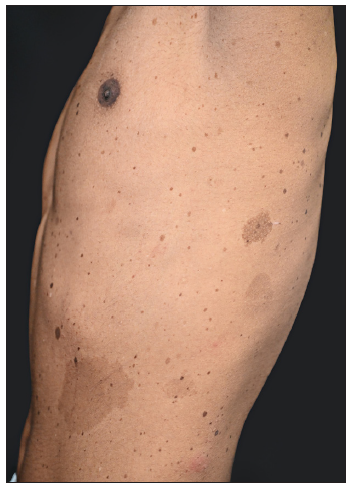
- Café-au-lait macules of varying sizes and numerous freckles distributed on the left chest.
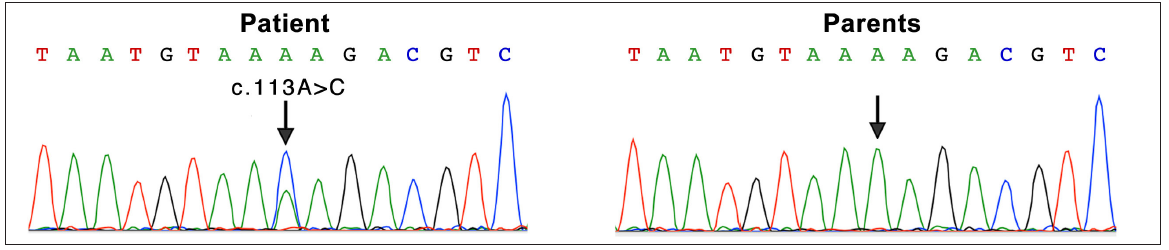
- Mutation analysis in the patient and his parents. A heterozygous mutation of c.113A>C (Arrow indicated) in exon 2 of the KITLG gene was detected in the patient. His parents didn’t carry this mutation (Arrow indicated corresponding nucleotide). A, Adenosine; G, Guanine; C, Cytosine; T, Thymine.
KITLG encodes the KIT receptor tyrosine kinase ligand which is locally expressed in epidermal keratinocytes and dermal endothelial cells in human skin.2 Following its interaction with the KIT receptor, the KIT signalling pathway is activated, leading to enhanced melanocyte proliferation and melanin synthesis.2 Variants in KITLG have been shown to contribute to a wide range of disorders, encompassing autosomal dominant conditions such as non-syndromic deafness (OMIM #616697) and FPHH (OMIM #145250), as well as autosomal recessively inherited Waardenburg syndrome type 2F (OMIM #619947). Recently, another autosomal recessive disease, hypomelanosis, and sensorineural hearing loss was identified as being caused by biallelic KITLG mutations.3 Distinct mutations may generate different consequences, such as loss or gain of function, or exhibit a dominant-negative effect on the wild-type allele. FPHH is associated with heterozygous gain-of-function variants in the KITLG gene.4 To date, there are only 10 KITLG mutations reported to cause FPHH [Figure 2b]. All the reported mutations affected the residues within the KIT ligand domain, leading to an increased affinity to the kit receptor.4 Most of the mutations (seven in nine) are in exon 2 of KITLG and the affected residues primarily reside within the conserved VTNNV region (residues 33–37).5 The other two mutations (Asp110Val and Glu113Lys) are located in exon 4 which encodes the core of the ligand that is crucial for binding and activating the receptor.1, 5 The mutation c.113A>C (p.Lys38Thr) reported in this study is a novel mutation which further expands the mutation spectrum and the hot-spot region of FPHH to include residues 33-38 (‘VTNNVK’). Interestingly, our patient exhibited a relatively large number of CALMs and lentigines which require differentiation from pigmentary diseases that also manifest with multiple CALMs and lentigines, such as neurofibromatosis type 1, legius syndrome, LEOPARD syndrome, and tuberous sclerosis complex. Further cases of FPHH caused by the same mutation are warranted to elucidate if the extensive involvement is mutation related.
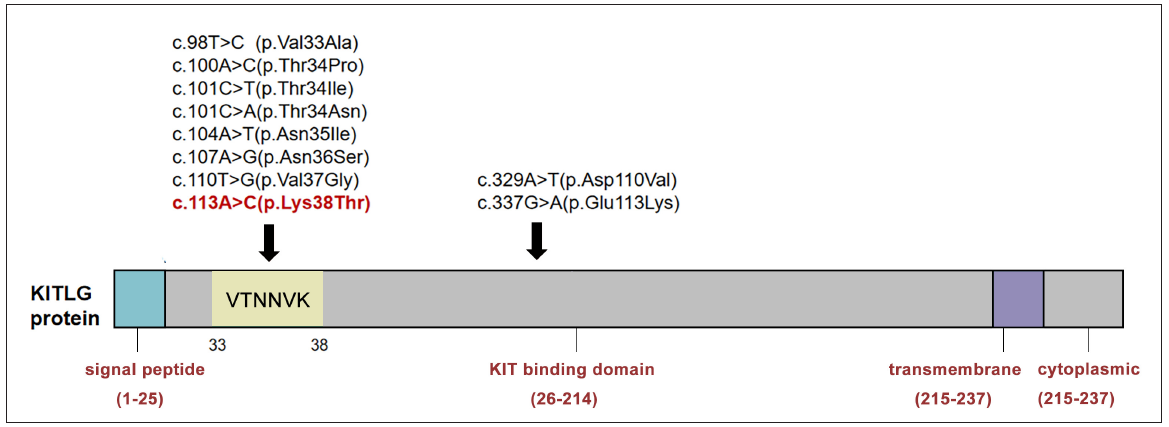
- Summary of FPHH-associated KITLG gene mutations. Most of these mutations are located in a hot mutation cluster ‘VTNNVK’ of the KIT-binding domain. The novel mutation (c.113A>C, p.Lys38Thr) reported in our study is marked in red. V, Val = Valine; Ala = Alanine; T, Thr = Threonine; Pro = Proline; Ile = Isoleucine; N, Asn = Asparagine; Ser = Serine; Gly = Glycine; K, Lys = Lysine; Asp = Aspartic acid; Glu = Glutamic acid.
Mutations in KIT, encoding for the receptor of KITLG, could lead to piebaldism (loss of function in KIT) or skin hyperpigmentation (gain of function in KIT). Occasionally, patients with piebaldism may exhibit multiple CALMs, while the hypopigmentation is accompanied by a distinctive white forelock.
Conclusion
In cases of skin hyperpigmentation that results from gain-of-function mutations in KIT, patients may develop gastrointestinal stromal tumours; however, hypomelanosis is not a concurrent feature.
Ethical approval
The research/study approved by the Institutional Review Board at Southern Medical University Dermatology Hospital Ethics Committee, number 2022035, dated Aug 17, 2023.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
References
- De novo mutation in KITLG gene causes a variant of familial progressive hyper- and hypo-pigmentation (FPHH) Mol Genet Genomic Med. 2021;9:e1841.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Gain-of-function mutation of KIT ligand on melanin synthesis causes familial progressive hyperpigmentation. Am J Hum Genet. 2009;84:672-7.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Biallelic KITLG variants lead to a distinct spectrum of hypomelanosis and sensorineural hearing loss. J Eur Acad Dermatol Venereol. 2022;36:1606-11.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- KITLG mutations cause familial progressive hyper- and hypopigmentation. J Invest Dermatol. 2011;131:1234-9.
- [CrossRef] [PubMed] [Google Scholar]
- Identification of a novel mutation in the KITLG gene in a Chinese family with familial progressive hyper- and hypopigmentation. BMC Med Genomics. 2021;14:12.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]




