
Translate this page into:
A rare case of infantile desmoid-type fibromatosis on the thigh
Corresponding author: Prof. Guiying Zhang, No. 139 Renmin Road, Changsha 410011, Hunan, China. zgylinda@csu.edu.cn
-
Received: ,
Accepted: ,
How to cite this article: Luo S, Tang G, Zhanga G. A rare case of infantile desmoid-type fibromatosis on the thigh. Indian J Dermatol Venereol Leprol 2021;87:601.
Desmoid-type fibromatosis is an uncommon group of fibrous neoplasms which originate from musculoaponeurotic tissues. It accounts for <3% of all soft-tissue tumors and 0.03% of all neoplasms.1 The annual incidence is about 2–4 cases/million with two peaks in 6–15 and 25–35-years-old.2 Desmoid-type fibromatosis occurring before ten years old is also called infantile fibromatosis which have the predilection sites in head, neck, thigh and shoulder.3 Herein, we described a rare case of infantile desmoid-type fibromatosis on the right thigh of an eight month boy.
An eight month-old Chinese boy presented with a dark red plaque on his right thigh at birth. This boy had a natural delivery without history of asphyxiation and birth trauma. There was no significant family history or genetic history. Because there were no other associated symptoms, this patient did not seek medical help eight months when the plaque enlarged gradually. Physical examination revealed a large dark red plaque on the root of his right thigh measuring 5.0 × 8.0 × 0.3 cm, and the surface of the lesion was rough with a red halo around it [Figure 1]. Ultrasonography showed soft-tissue mass involving the dermis and the subcutaneous tissue. Skin biopsy showed a diffused dense infiltration of spindle cells with round or spindle nuclei, arranged in fascicular and storiform pattern [Figure 2a]. Mitotic figures were occasionally seen in high power field without atypicality. At the periphery of the lesion, lymphocytes, histiocytes and occasional multinucleated giant cells infiltration were seen [Figure 2b]. Immunohistochemical staining showed positive for β-catenin [Figure 2c] and negative for desmin, smooth muscle actin and CD34. These findings suggested a diagnosis of desmoid-type fibromatosis. Subsequently, the patient had an extended excision of the skin lesion with negative margins. Post-operative monitoring and magnetic resonance imaging (MRI) scan did not show evidence of recurrence after two years follow-up.
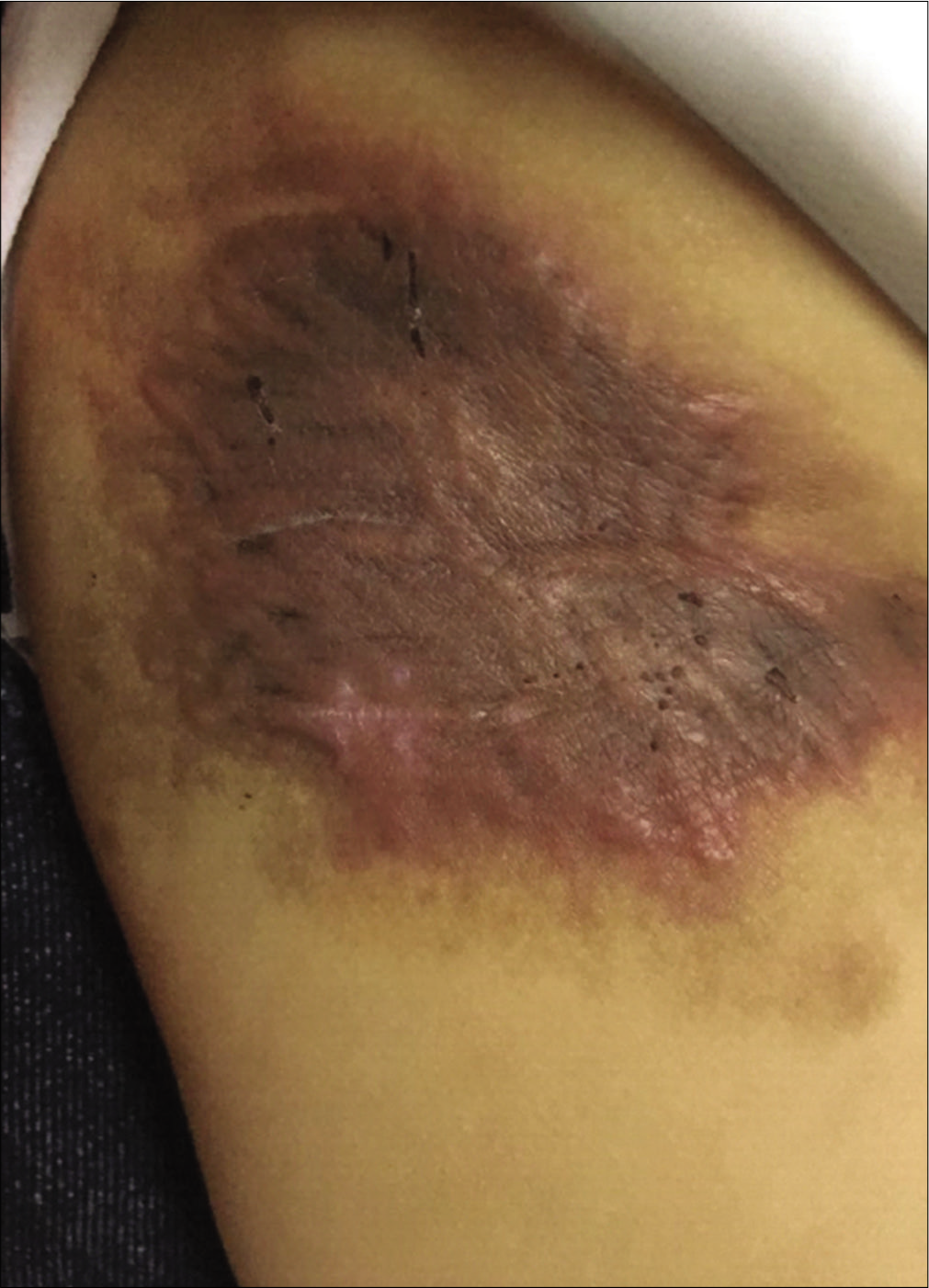
- Clinical features: A dark red plaque on the right thigh
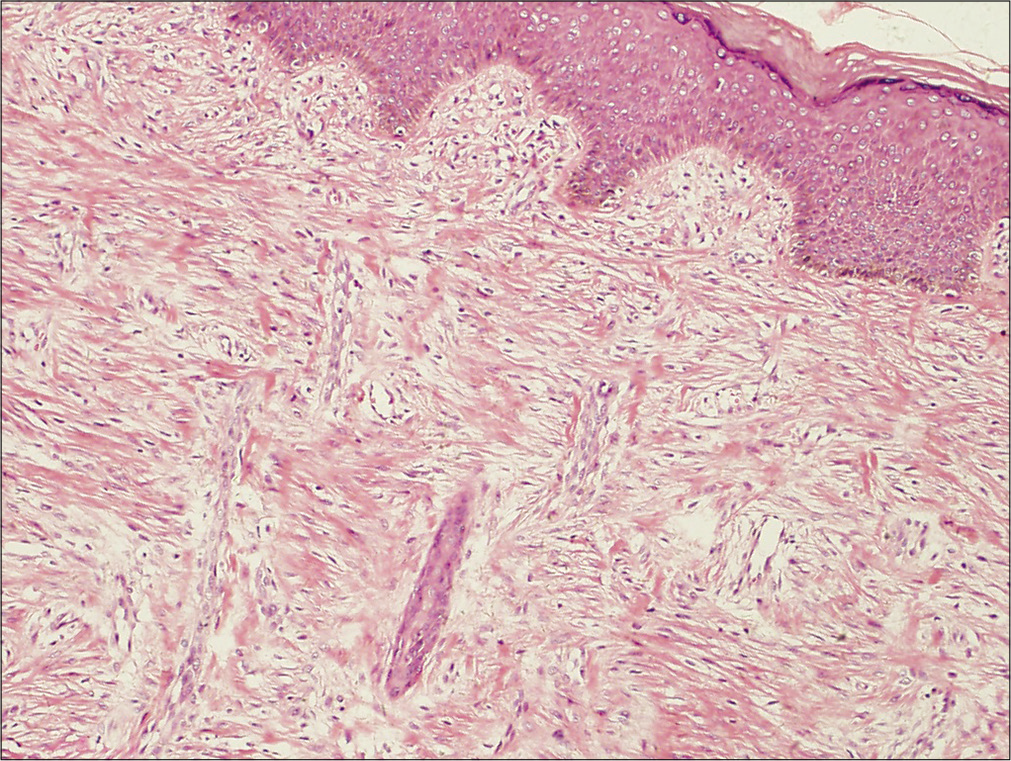
- Histological findings of the tumor. The tumor cells are in fascicular and storiform pattern without capsule at low-power view (H and E, ×100)
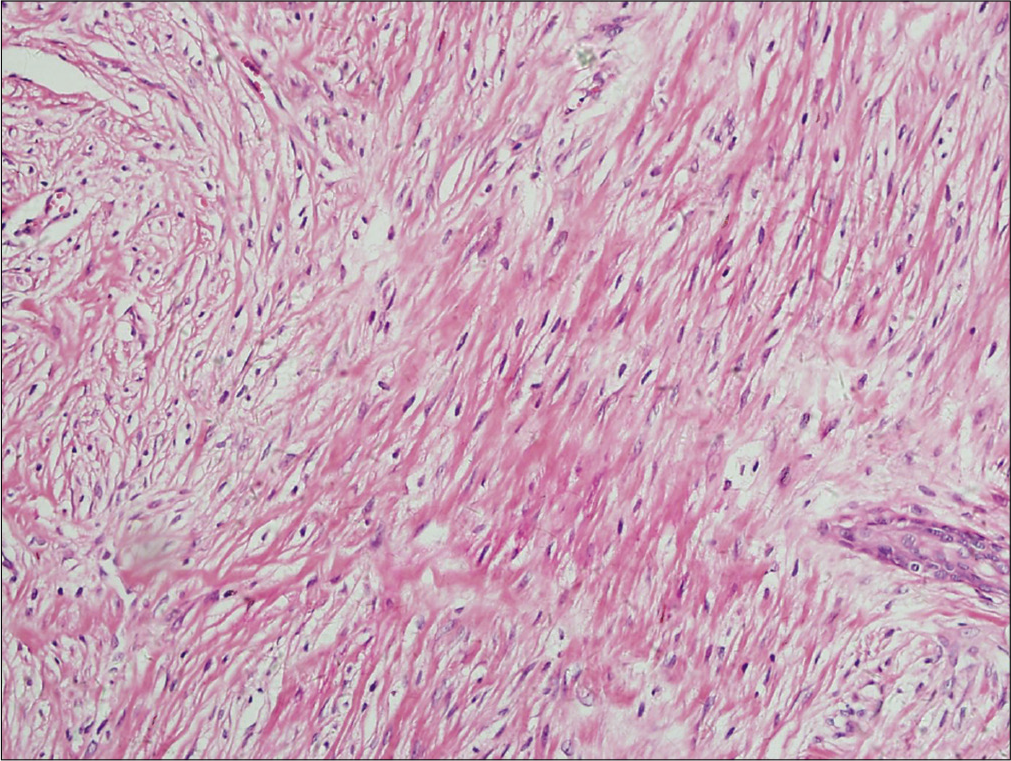
- Histological findings of the tumor. The tumor is composed of an admixture of plump spindle cells with round or spindle nuclei in a background of variably hyalinized and myxoid collagenous stroma with occasional mitotic figures at high-power view (H and E, ×400)
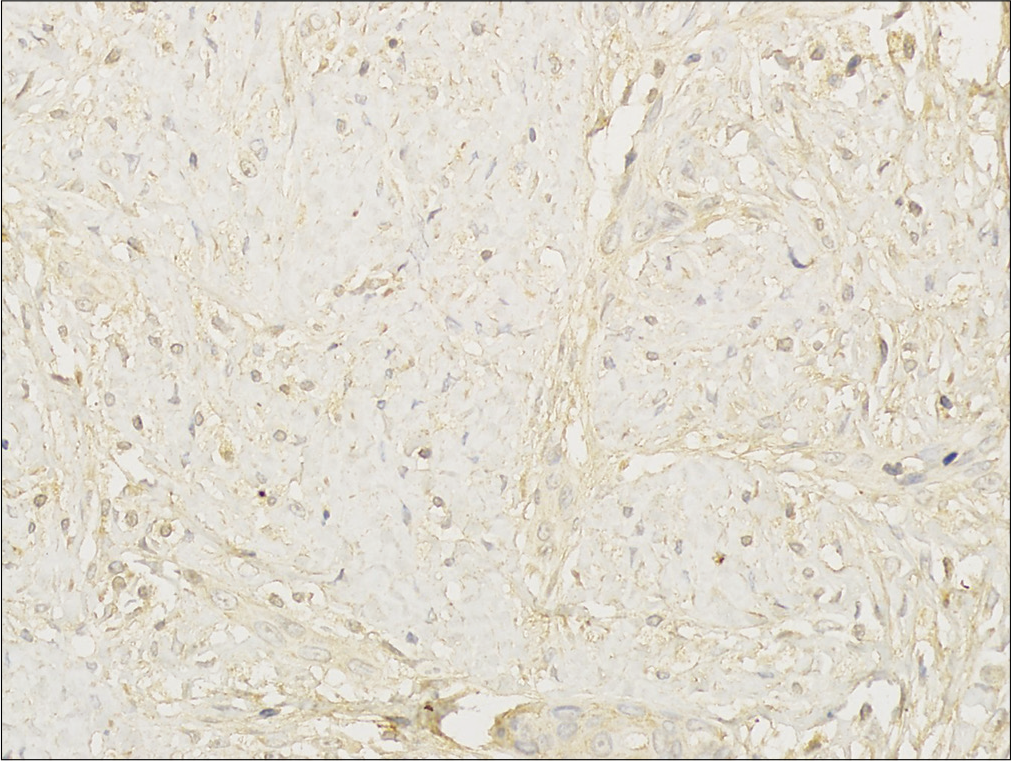
- Histological findings of the tumor. Immunohistochemical staining stain shows that the tumor cells are positive for β-catenin (β-catenin staining, ×400)
Desmoid-type fibromatosis was first named by Muller in year of 1838 and regarded as a subtype of locally aggressive fibromatosis which is characterized by nonmetastasis, infiltrative growth and local recurrence.1 These unique characteristics make it a challenge in clinical diagnosis and management.1 Multiple factors including genetic abnormalities, abnormal growth regulation of connective tissue and endocrine have been confirmed to play roles in the pathogenesis of desmoid-type fibromatosis.4 Molecular studies have found that mutations in CTNNB1, the gene encoding β-catenin, are seen in approximately 85% of sporadic cases and overactivation of the Wnt/APC/β-catenin pathway contributes to neoplasia and development of fibrous tissue.5
Desmoid-type fibromatosis generally presents with a slowly growing skin lump with or without pain.6 Biological behavior is often unpredictable although some lesions may behave more aggressively. Desmoid-type fibromatosis was rarely reported to metastasize or undergo sarcomatous transformation, but local spontaneous recurrence is common.6 The histopathological features of desmoid-type fibromatosis are characterized by proliferation of uniformly spindle cells with abundant collagen. Immature mesenchymal-like cells and fibroid hyperplasia can be seen with minimal nuclear atypia.7,8 Immunohistochemical stains are helpful and should be perform in every case. Immunohistochemical staining usually show positive for actin and negative or focally positive for desmin. Positive β-catenin in nuclear staining would be helpful in diagnosis.7,8
Several fibrous tumors should be considered in the differential diagnosis of desmoid-type fibromatosis, especially fibrosarcoma and low-grade myofibroblastic sarcoma. Fibrosarcoma is a malignant tumor characterized by prominent abnormal mitotic activity and typically a herring-bone pattern. Low-grade myofibroblastic sarcoma is a rare type of malignant myofibroblastic tumor which composed of fascicles of cells with vesicular nuclei and indistinct cytoplasmic margins. Fibrosarcoma and low-grade myofibroblastic sarcoma tend to be negative for β-catenin. In some cases, multicentric desmoid-type fibromatosis may be skin feature of Gardner’s syndrome.9 Gardner’s syndrome is an inherited condition also accompanied with familial adenomatous polyposis, and CTNNB1 sequencing might be helpful to screen and identify some difficult cases in children.10
In terms of treatment, expanded resection is the first choice.2,3,6,11 Microscopic positive margins may be acceptable if achieving negative margins would result in greater morbidity. If incomplete resection is performed, the local recurrence rate could be high up to 65%.2,3 MRI scan is helpful to decide the boundaries of the tumor. Recurrence is most likely happening in the 1st year, so long-term clinical and MRI follow-up after surgery are recommended in pediatric patients for early detection of tumor recurrence.3,4,11
In summary, desmoid-type fibromatosis is a rare type of fibrous tumors which usually manifests as an ignored fast-growing skin plaque and characterized by benign mesenchymal-like tumor with fibrous hyperplasia. Therefore, it is suggested that clinicians should carefully identify such cutaneous tumor and earlier conduct biopsy to avoid wrong diagnosis.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- The evolving classification of soft tissue tumours-an update based on the new 2013 WHO classification. Histopathology. 2014;64:2-11.
- [CrossRef] [PubMed] [Google Scholar]
- Desmoid-type fibromatosis: Who, when, and how to treat. Curr Treat Options Oncol. 2017;18:29.
- [CrossRef] [PubMed] [Google Scholar]
- Current update on desmoid fibromatosis. J Comput Assist Tomogr. 2019;43:29-38.
- [CrossRef] [PubMed] [Google Scholar]
- The role of APC and beta-catenin in the aetiology of aggressive fibromatosis (desmoid tumors) Eur J Surg Oncol. 2009;35:3-10.
- [CrossRef] [PubMed] [Google Scholar]
- Infantile fibromatosis: A case report and review of the literature. Br J Oral Maxillofac Surg. 2011;49:e30-2.
- [CrossRef] [PubMed] [Google Scholar]
- Specific mutations in the beta-catenin gene (CTNNB1) correlate with local recurrence in sporadic desmoid tumors. Am J Pathol. 2008;173:1518-27.
- [CrossRef] [PubMed] [Google Scholar]
- Immunohistochemistry for beta-catenin in the differential diagnosis of spindle cell lesions: Analysis of a series and review of the literature. Histopathology. 2007;51:509-14.
- [CrossRef] [PubMed] [Google Scholar]
- Desmoid tumors in familial adenomatous polyposis. Anticancer Res. 2017;37:3357-66.
- [CrossRef] [PubMed] [Google Scholar]
- CTNNB1 mutation analysis is a useful tool for the diagnosis of desmoid tumors: A study of 260 desmoid tumors and 191 potential morphologic mimics. Mod Pathol. 2012;25:1551-8.
- [CrossRef] [PubMed] [Google Scholar]
- Biology and treatment of aggressive fibromatosis or desmoid tumor. Mayo Clin Proc. 2017;92:947-64.
- [CrossRef] [PubMed] [Google Scholar]




